Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Wydział Nadzoru Rynku

2
Wydział Nadzoru Rynku W pierwszym okresie działalności Urzędu – 3 osoby Następnie – 4 osoby /1 osoba z Wydziału Rejestracji/ 0d połowy 2004 r. – 6 osób /połączenie z samodzielnym stanowiskiem ds. incydentów medycznych - 1 osoba + 1 osoba z Wydziału Rejestracji/ Od lutego 2005 r. - 9 osób /połączenie z samodzielnym stanowiskiem ds. jednostki notyfikowanej – 2 osoby + 1 osoba nowo przyjęta/

3
Wydział Nadzoru Rynku 6 inżynierów 1 mgr chemii 1 mgr farmacji
1 mgr matematyki / w tym dwóch audytorów/

4
Nadzór nad wyrobami medycznymi
Kontrola podmiotów odpowiedzialnych za wprowadzanie do obrotu i do używania Nadzór nad incydentami medycznymi Informacja nt. wyrobów medycznych, interpretacja przepisów

5
Podmioty odpowiedzialne za wprowadzanie do obrotu i do używania
Wytwórca Autoryzowany przedstawiciel Importer Dystrybutor [w przypadku informacji o nieprawidłowościach] Podmiot odpowiedzialny za wprowadzenie do obrotu wyrobu medycznego

6
Ustawa z dnia 20 kwietnia 2004 r. o wyrobach medycznych
Podstawy prawne Ustawa z dnia 20 kwietnia 2004 r. o wyrobach medycznych wdrażająca postanowienia dyrektyw 90/385/EEC o aktywnych implantach 93/42/EEC o wyrobach medycznych ze zmianami w dyrektywie 98/79/EC i uzupełnieniami z: dyrektywy 2003/12/EC o implantach piersi dyrektywy 2003/32/EC o wyrobach medycznych z tkankami pochodzenia zwierzęcego dyrektywy 2000/70/EC o wyrobach medycznych z krwią lub osoczem oraz dyrektywy 2001/104/EC o okresach przejściowych dla w/w wyrobów 98/79/EC – wyroby do diagnostyki in vitro Ustawa o Urzędzie Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych

7
Rozporządzenia Rozporządzenie Ministra Zdrowia z dnia 30 kwietnia 2004 r. w sprawie klasyfikacji wyrobów medycznych do różnego przeznaczenia Rozporządzenie Ministra Zdrowia z dnia 3 listopada 2004 r. w sprawie wymagań zasadniczych dla wyrobów medycznych do różnego przeznaczenia Rozporządzenie Ministra Zdrowia z dnia 3 listopada 2004 r. w sprawie wymagań zasadniczych dla wyrobów medycznych do implantacji Rozporządzenie Ministra Zdrowia z dnia 3 listopada 2004 r. w sprawie wymagań zasadniczych dla wyrobów medycznych do diagnostyki in vitro Rozporządzenie Ministra Zdrowia z dnia 30 kwietnia 2004 r. w sprawie sposobu zgłaszania incydentów medycznych oraz dalszego postępowania po ich zgłoszeniu

8
Procedury Wydziału Kontrola podmiotów odpowiedzialnych
Nadzór nad incydentami Współpraca z organami inspekcji Wstrzymywanie, ograniczanie, wycofywanie w obrocie i w używaniu Postępowanie w przypadku reklamacji jakościowych

9
Wydział Nadzoru Rynku Kontrola wytwórców w zakresie spełnienia wymagań ustawy: w zakresie projektowania; wytwarzania; pakowania; oznakowywania; przechowywania; przetwarzania; przeprowadzania remontu odtworzeniowego; wystawiania; nadawania przewidzianego zastosowania; sterylizacji; zestawiania systemów lub zestawów; wprowadzania do obrotu i do używania na terenie RP

10
Wydział Nadzoru Rynku Kontrola autoryzowanych przedstawicieli / importerów w zakresie spełnienia wymagań ustawy: dokumentacji, która powinni otrzymać od wytwórcy; umowy autoryzacyjnej lub na import; oznakowywania; przechowywania; wystawiania; wprowadzania do obrotu i do używania na terenie RP

11
Wydział Nadzoru Rynku Kontrola innych podmiotów odpowiedzialnych
w zakresie pakowania; oznakowywania; przechowywania; wystawiania; sterylizacji; zestawiania systemów lub zestawów; wprowadzania do obrotu i do używania na terenie RP

12
Wprowadzanie do obrotu i do używania
Do obrotu i do używania mogą być wprowadzane wyroby medyczne spełniające wymagania określone w ustawie (art. 4.1.) Do obrotu i do używania mogą być wprowadzane wyroby medyczne oznakowane znakiem CE (art. 5.1.) i dla których przeprowadzono ocenę zgodności Wyrób medyczny wprowadzany do obrotu i do używania musi spełniać określone dla niego wymagania zasadnicze (art. 16.1)
 Do obrotu i do używania mogą być wprowadzane wyroby medyczne oznakowane znakiem CE (art. 5.1.) i dla których przeprowadzono ocenę zgodności. Wyrób medyczny wprowadzany do obrotu i do używania musi spełniać określone dla niego wymagania zasadnicze (art. 16.1)")
13
Wyrób medyczny O medycznym przeznaczeniu wyrobu decyduje wytwórca; musi być to zgodne z ustawową definicją Musi to uzasadnić przeprowadzając postępowanie mające na celu potwierdzenie bezpieczeństwa wyrobu i skuteczności działania Musi prawidłowo sklasyfikować wyrób i przeprowadzić ocenę zgodności (w przypadku wyrobów klasy I z funkcją pomiarową lub jałowych, wyrobów klasy IIa, IIb i III przy udziale jednostki notyfikowanej) Musi również wyrób właściwie oznakować, załączyć instrukcję użycia sformułowaną tak, aby była jasna dla każdego użytkownika
 Musi również wyrób właściwie oznakować, załączyć instrukcję użycia sformułowaną tak, aby była jasna dla każdego użytkownika.")
14
Wyrób medyczny Wyrób medyczny musi spełniać definicję ustawową tzn. służyć do: diagnozowania, zapobiegania, monitorowania, leczenia, łagodzenia chorób diagnozowania, zapobiegania, monitorowania, leczenia, łagodzenia lub kompensowania upośledzeń badania, zastępowania lub modyfikowania budowy anatomicznej lub procesu fizjologicznego regulacji poczęć

15
Klasyfikacja - zasady ogólne
Czas kontaktu Miejsce kontaktu Inwazyjność Aktywność

16
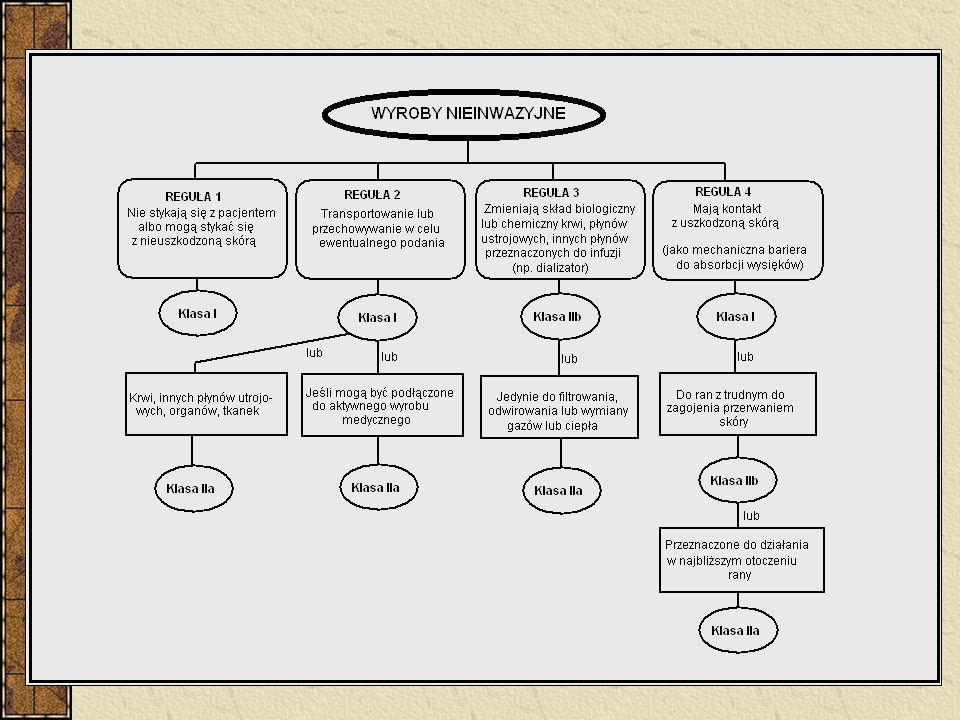
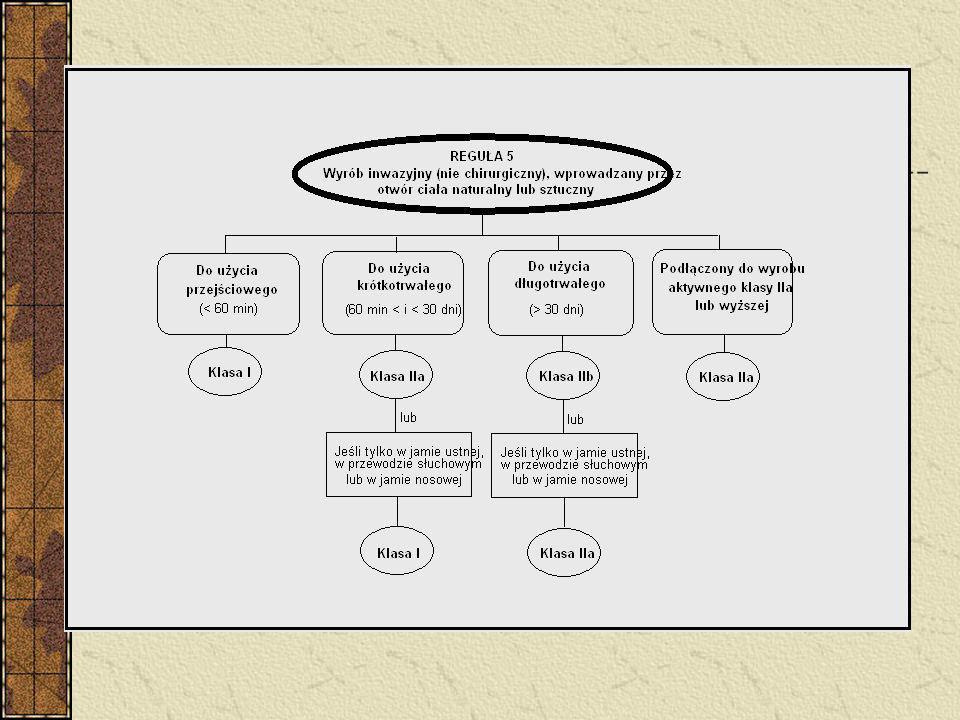
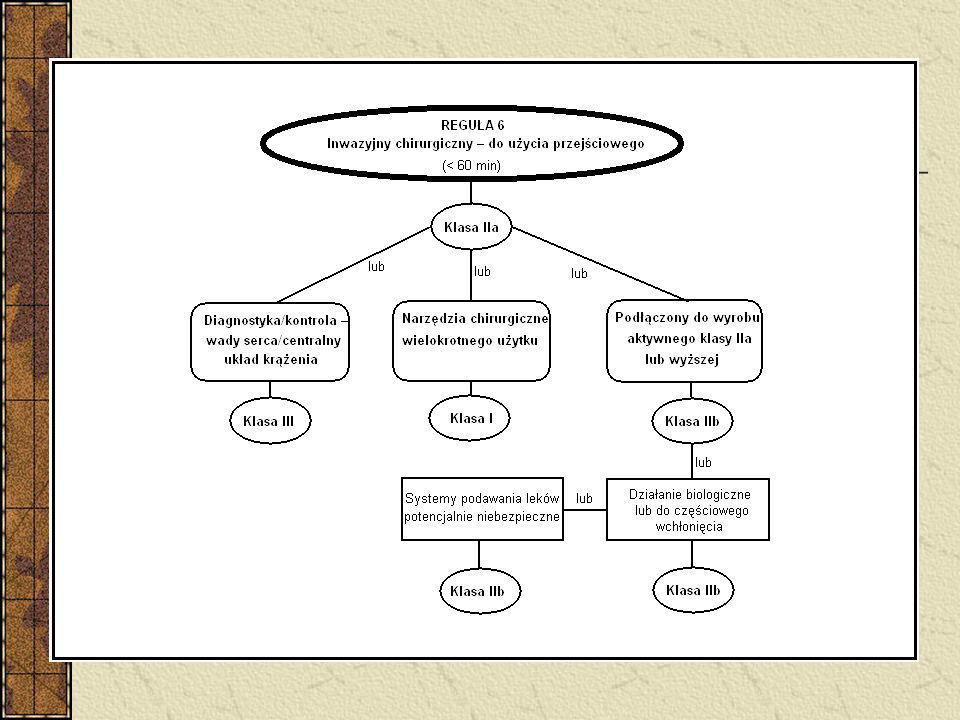
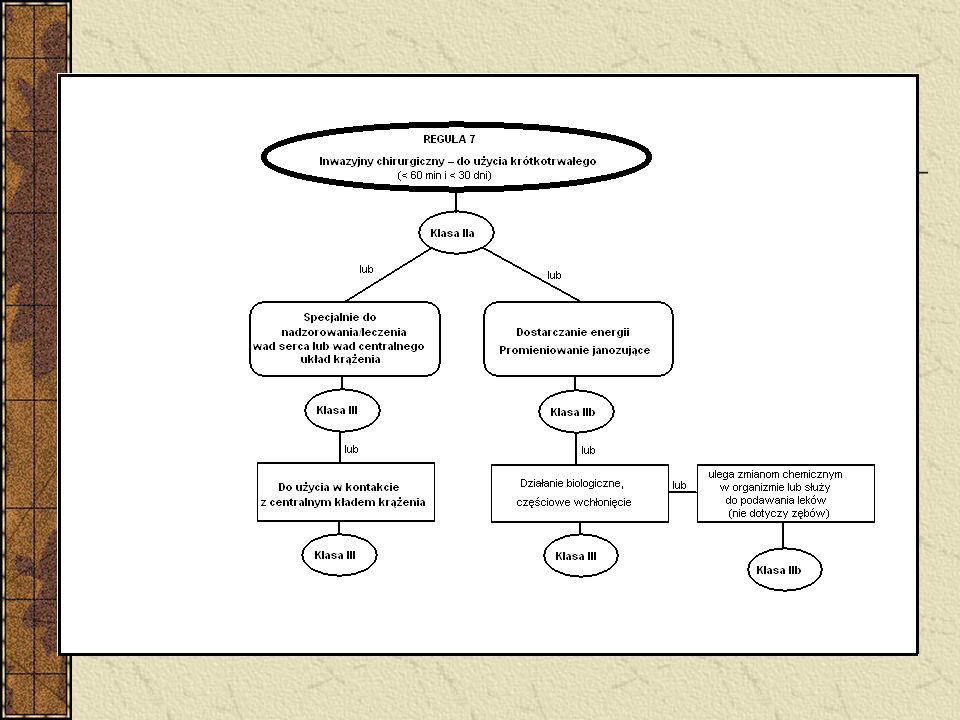
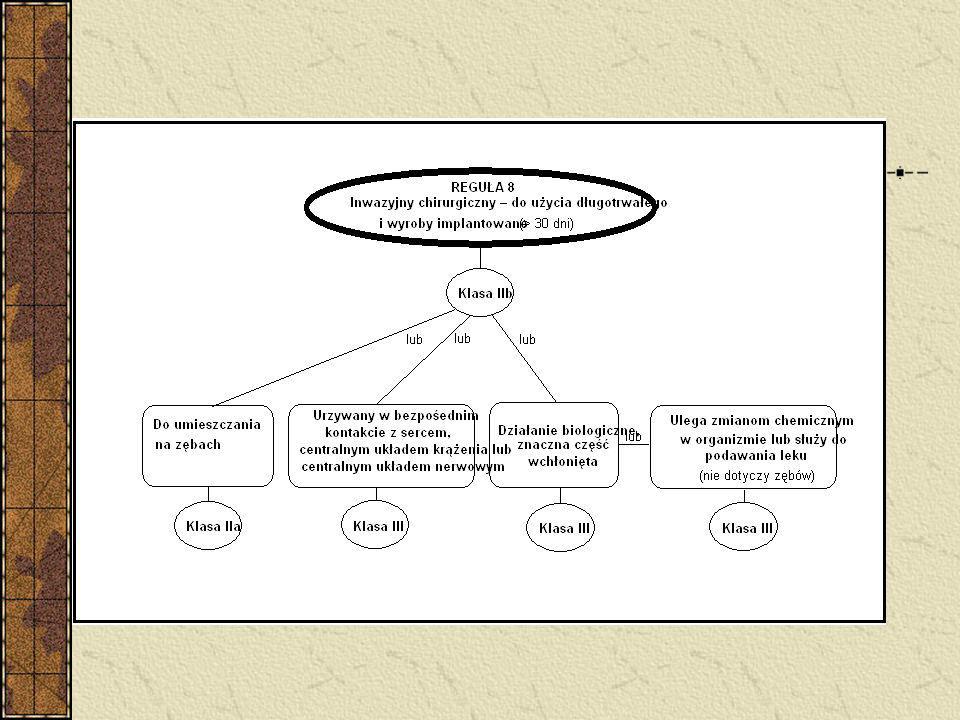
Klasyfikacja Wyroby nieinwazyjne – 4 reguły (1, 2, 3, 4)
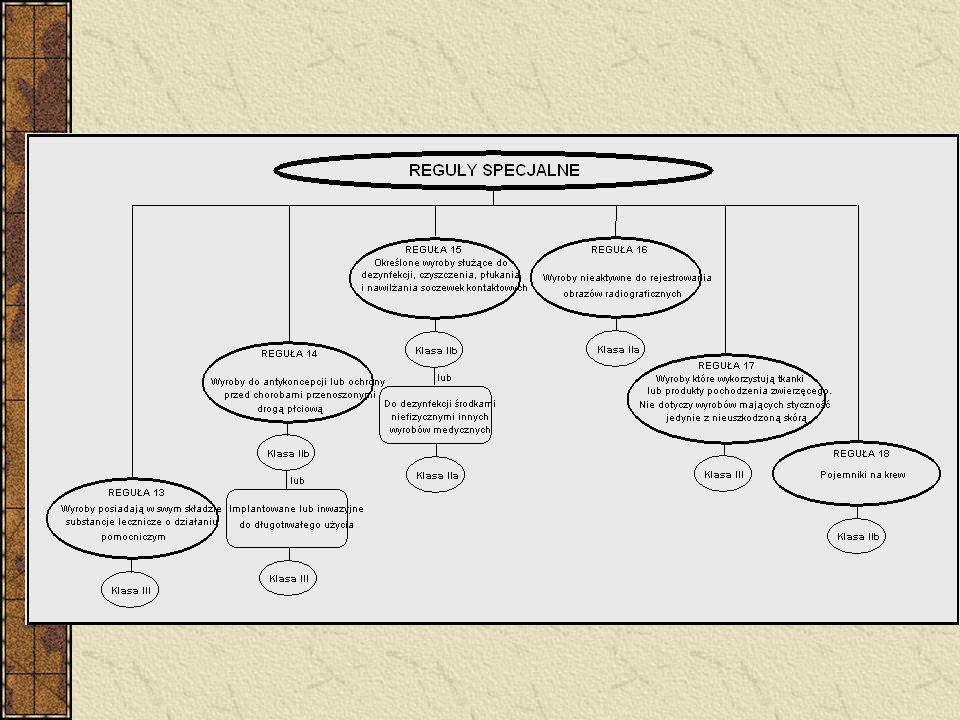
Wyroby inwazyjne – 4 reguły (5, 6, 7, 8) Wyroby aktywne – 4 reguły (9, 10, 11, 12) 6 reguł specjalnych - wyroby zawierające jako integralną część substancję uznaną z produkt leczniczy - wyroby używane w celach antykoncepcji lub zapobiegania rozprzestrzenianiu się chorób przenoszonych drogą płciową - wyroby przeznaczone do dezynfekcji - nieaktywne wyroby do rejestracji diagnostycznych obrazów rentgenowskich - wyroby wyprodukowane z wykorzystaniem pozbawionych zdolności do życia tkanek zwierzęcych - worki na krew Implanty piersi – klasa III
 Wyroby aktywne – 4 reguły (9, 10, 11, 12) 6 reguł specjalnych. - wyroby zawierające jako integralną część substancję uznaną z produkt leczniczy. - wyroby używane w celach antykoncepcji lub zapobiegania rozprzestrzenianiu się chorób przenoszonych drogą płciową. - wyroby przeznaczone do dezynfekcji. - nieaktywne wyroby do rejestracji diagnostycznych obrazów rentgenowskich. - wyroby wyprodukowane z wykorzystaniem pozbawionych zdolności do życia tkanek zwierzęcych. - worki na krew. Implanty piersi – klasa III.")
22
Gdy są użuwane do monitorowania procesów fizjologicznych, których
zmiany mogłyby spowodować natychmiastowe zagrożenie życia

24
Określa załącznik nr 1 do rozporządzenia Ministra Zdrowia
Wymagania zasadnicze Określa załącznik nr 1 do rozporządzenia Ministra Zdrowia Wymagania ogólne – dotyczą projektowania i wytwarzania, parametrów działania, opakowania Wymagania dotyczące projektu i wykonania obejmują zapewnienie dobrych właściwości chemicznych, fizycznych i biologicznych; właściwy dobór materiałów zwłaszcza pod względem biozgodności; właściwe zabezpieczenie przed infekcją i skażeniem biologicznym; właściwy dobór parametrów konstrukcyjnych i oddziaływania środowiska; w przypadku wyrobów z funkcją pomiarową – dokładność i stałość w granicach tolerancji; zapewnienie ochrony przed promieniowaniem; określone są także wymagania dla wyrobów z zewnętrznym źródłem zasilania lub w takie źródło wyposażonych Informacje dostarczane przez wytwórcę

25
Procedury oceny zgodności (1)
")
26
Procedury oceny zgodności (2)
")
27
Procedury oceny zgodności (3)
")
28
Procedury oceny zgodności (4)
Klasa III + aktywne implanty Załącznik nr 3 + Załącznik nr 4 Załącznik nr 2 Załącznik nr 3 + Załącznik nr 5 Pełny system jakości z oceną projektu Ocena typu + Weryfikacja EC Ocena typu + Zapewnienie jakości produkcji CE CE CE

29
Klasyfikacja W przypadku nieprawidłowej klasyfikacji lub kwalifikacji wytwórca jest zobowiązany do jej ponownego przeprowadzenia W przypadku rozbieżności co klasyfikacji lub kwalifikacji pomiędzy wytwórcą a jednostką notyfikowaną decyzję podejmuje Prezes

30
Deklaracja zgodności Dane wytwórcy /nazwa i adres/
Dane autoryzowanego przedstawiciela /jeżeli dotyczy/ Dane wyrobu Oświadczenie o zgodności wyrobu z odpowiednimi wymaganiami rozporządzenia / oświadczenie o zgodności z wymaganiami zasadniczymi wg załącznika 1 oraz wg którego załącznika lub załączników przeprowadzono ocenę zgodności/ Powołanie norm zharmonizowanych lub specyfikacji Numer jednostki notyfikowanej, jeżeli brała udział w ocenie zgodności Deklaracja musi być podpisana przez osobę upoważnioną przez wytwórcę oraz oznakowana datą i miejscem wystawienia.

31
Działania pokontrolne
W przypadku stwierdzenia uchybień wydawane są zalecenia pokontrolne i określany termin ich wykonania. Nie zastosowanie się w terminie do wykonania zaleceń może skutkować decyzją Prezesa o wstrzymaniu, ograniczeniu lub wycofaniu wyrobów zarówno w obrocie jak i w używaniu.

32
Uprawnienia Prezesa W przypadku uzyskania informacji, że wyrób nie spełnia wymagań: może żądać udostępnienia próbek do przeprowadzenia badań i weryfikacji wyrobu zlecić przeprowadzenie badania w jednostce posiadającej odpowiednie wyposażenie badawcze i kwalifikowany personel Jeżeli wyniki badań i weryfikacji potwierdzą, że wyrób nie spełnia określonych wymagań koszty ponosi podmiot odpowiedzialny, a Prezes może wydać odpowiednią w sprawie decyzję o wydaniu notatki doradczej z podaniem informacji o dalszym postępowaniu w sprawie.

33
Uprawnienia Prezesa O podjętych decyzjach:
wstrzymania wprowadzania do obrotu i do używania ograniczenia w obrocie lub w używaniu wycofania z obrotu wycofania z obrotu i z używania Prezes powiadamia Komisję Europejską i organy kompetentne państw członkowskich UE i EFTA podając uzasadnienie tych decyzji.

34
Przepisy karne Art. 78 – dotyczy wprowadzania wyrobów medycznych do obrotu i do używania bez przeprowadzenia oceny zgodności Art dotyczy wprowadzania do obrotu i do używania systemów lub zestawów albo sterylizacji takich systemów lub zestawów albo wyrobów oznakowanych CE bez spełnienia warunków ustawy Art. 81 – dotyczy wprowadzania na terytorium RP wyrobów bez oznakowania w języku polskim Art. 82 – dotyczy znakowania CE /także z numerem jednostki notyfikowanej/ bez spełnienia warunków ustawy lub innego znakowania wprowadzającego w błąd Art. 83 – dotyczy wprowadzania do obrotu i do używania wyrobów przeterminowanych lub wyrobów których oznakowanie może wprowadzić w błąd co do właściwości lub działania Art. 85 – dotyczy nie zgłoszenia incydentu medycznego Art. 86 – dotyczy uniemożliwiania lub utrudniania przeprowadzenia kontroli

35
Wydział Nadzoru Rynku (I)
Kontrole wytwórców W 2003 r. – przeprowadzono 3 kontrole W 2004 r. – 35 kontroli Od stycznia do marca 2005 r. – 19 kontroli

36
Wydział Nadzoru Rynku (II)
Informacja o wyrobach medycznych 01/10/2004 – 31/12/2004 Udzielono odpowiedzi w 30 sprawach 01/01/2005 – 31/03/2005 Udzielono odpowiedzi również w 30 sprawach

37
Wydział Nadzoru Rynku (III)
Udzielanie odpowiedzi na pytania problemowe dotyczące przepisów, kwalifikacji, klasyfikacji. Rozpatrywanie spraw skierowanych z Wydziału Rejestracji. Opracowywania na potrzeby służb celnych, policji, prokuratury, organów inspekcji, ministerstwa.

Podobne prezentacje














. Urząd.>")
>")



