Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Hematopatologia

2
Układ czerwonokrwinkowy
Anemia Z utraty krwi: ostra, przewlekła Spadek produkcji: defekt dojrzewania (megaloblastyczna, niedobór żelaza, MDS), zaburzenia proliferacji (defekt komórek pnia, wyparcie hemopoezy, anemia chorób przewlekłych) Hemolityczna: zaburzenia struktury krwinek – hemoglobinopatie (thalassemia, anemia sierpowata), nieprawidłowości błony (wrodzona sferocytoza, eliptocytoza), nieprawidłowości enzymatyczne (niedobór dehydrogenzy glukozo-6-fosforanowej), PNH (defekty cząstek łączących się z fosfatydyloinozytolem – genetyczny defekt nabyty) Czynniki zewnętrzne – AIHA (ciepłe i zimne p/ciała), mikroangiopatyczna (m.in. HUS), pasożytnicze (malaria, babesioza) Erytrocytoza
, zaburzenia proliferacji (defekt komórek pnia, wyparcie hemopoezy, anemia chorób przewlekłych) Hemolityczna: zaburzenia struktury krwinek – hemoglobinopatie (thalassemia, anemia sierpowata), nieprawidłowości błony (wrodzona sferocytoza, eliptocytoza), nieprawidłowości enzymatyczne (niedobór dehydrogenzy glukozo-6-fosforanowej), PNH (defekty cząstek łączących się z fosfatydyloinozytolem – genetyczny defekt nabyty) Czynniki zewnętrzne – AIHA (ciepłe i zimne p/ciała), mikroangiopatyczna (m.in. HUS), pasożytnicze (malaria, babesioza) Erytrocytoza.")
3
Anemia = spadek Hb Normo-/mikro-/makrocytarna
Normo-/hipo-/hiperchromiczna

4
Anemia makrocytarna Noworodki, alkoholizm, niedobór kw. foliowego, B12, choroby wątroby, niedoczynność tarczycy, MDS, AA, HCL, ostre białaczki, leki: cyklofosfamid, metotreksat, antywirusowe, hipoglikemiczne (metformina), p/drgawkowe (fenytoina), p/zapalne (sulfasalazyna) Megaloblastyczna (B12) MCV zwykle powyżej 115; pancytopenia; 10 dni od leczenia – wzrost Hb; do normy – 8 tygodni; możliwe zaburzenia neurologiczne; wzrost ryzyka raka żołądka i NET żołądka (anemia złośliwa)
, p/drgawkowe (fenytoina), p/zapalne (sulfasalazyna) Megaloblastyczna (B12) MCV zwykle powyżej 115; pancytopenia; 10 dni od leczenia – wzrost Hb; do normy – 8 tygodni; możliwe zaburzenia neurologiczne; wzrost ryzyka raka żołądka i NET żołądka (anemia złośliwa)")
5
Anemia mikrocytarna Niedobór Fe, thalassemia, choroby przewlekłe, anemia sideroblastyczna, niedobór Cu, zatrucie Zn Z niedoboru Fe – I miejsce na świecie – najczęściej: kobiety (wiek rozrodczy i w ciąży), wcześniaki; słaba korelacja między objawami a poziomem Hb (inne enzymy z Fe); wczesny możliwy objaw – pica – patologiczne „smaki” (na niejadalne); wzrost Hb po 2 tygodniach, norma po 2 miesiącach; u starszych – kał na krew utajoną, gastro-/kolonoskopia
, wcześniaki; słaba korelacja między objawami a poziomem Hb (inne enzymy z Fe); wczesny możliwy objaw – pica – patologiczne „smaki (na niejadalne); wzrost Hb po 2 tygodniach, norma po 2 miesiącach; u starszych – kał na krew utajoną, gastro-/kolonoskopia.")
6
Anemia sierpowata Przewlekła anemia hemolityczna + okluzja mikrokrążenia – polimeryzacja odtlenowanej HB S; Większa wrażliwość na infekcję parwowirusem B19 – kryza aplastyczna; Zawały śledziony, martwice kości, ryzyko udaru, dysfunkcja nerek, żółtaczka, kamica żółciowa

7
Thalassemia Hipochromiczna, mikrocytarna anemia; spadek produkcji łańcuchów globiny alfa lub beta Deformacje szkieletu, hepatosplenomegalia, wtórna hemochromatoza

8
AIHA Ciepłe – IgG (37oC); częściej; pierwotna (idiopatyczna), wtórna – choroby autoimmunologiczne, limfoproliferacje, leki; splenomegalia (eliminacja w śledzionie); każdy wiek, K:M = 2:1; Zimne – IgM aglutyniny; pierwotna – przewlekła, głównie kobiety w starszym wieku; wtórna – głównie limfoproliferacje – m.in. makroglobulinemia Waldenstroema; infekcje – Mycoplasma pneumoniae, mononukleoza – zwykle samoistnie zanika w ciągu kilka tygodni; rzadko organomegalia
; częściej; pierwotna (idiopatyczna), wtórna – choroby autoimmunologiczne, limfoproliferacje, leki; splenomegalia (eliminacja w śledzionie); każdy wiek, K:M = 2:1; Zimne – IgM aglutyniny; pierwotna – przewlekła, głównie kobiety w starszym wieku; wtórna – głównie limfoproliferacje – m.in. makroglobulinemia Waldenstroema; infekcje – Mycoplasma pneumoniae, mononukleoza – zwykle samoistnie zanika w ciągu kilka tygodni; rzadko organomegalia.")
9
Erytrocytoza Spadek objętości osocza (względna)
Wzrost masy erytrocytów – policytemia: Pierwotna: rodzinna, PV Wtórna: choroby serca/płuc, duża wysokość n.p.m.; obniżone uwalnianie tlenu z Hb (wrodzone warianty o wysokim powinowactwie do tlenu, karboksyHb u palaczy); ektopowa produkcja EPO
; ektopowa produkcja EPO.")
10
Leukocytoza/leukopenia
Leukocytoza reaktywna – limfocytarna przy infekcjach wirusowych; reakcje białaczkowe; poliklonalna limfocytoza u palaczek (HLA-DR7); Leukopenia – polekowa, przy chorobach autoimmunologicznych
; Leukopenia – polekowa, przy chorobach autoimmunologicznych.")
11
Płytki Trombocytoza (możliwe – powikłania krwotoczne, zakrzepowe lub – bezobjawowo): Reaktywna – niedobór żelaza, nowotwory, zapalenia, infekcje, po splenektomii, po odstawieniu leków mielosupresyjnych Nowotworowe – TE, PV, PMF, CML (może być w pewnych postaciach AML – inv3) Trombocytopenia – ITP, DIC, indukowana heparyną, zakrzepowe (TTP, HUS) Skazy osoczowe, naczyniowe (główne cechy; hemofilia A i B)
 Trombocytopenia – ITP, DIC, indukowana heparyną, zakrzepowe (TTP, HUS) Skazy osoczowe, naczyniowe (główne cechy; hemofilia A i B)")
12
M. l. 23 Nigdy nie chorował; Po nacięciu ropnia zęba; 1 tydzień; zgon;
Przedłużające się krwawienie z dziąsła; złe samopoczucie; przejściowe duszności; drętwienie ręki; wymioty Przyjęty na dobę przed zgonem – anemia, trombocytopenia (15 000); narastające enzymy wątrobowe (ASPAT 2000 – 4000 w ciągu 24godzin)
; narastające enzymy wątrobowe (ASPAT 2000 – 4000 w ciągu 24godzin)")
13
Nowotwory układu krwiotwórczego
1. Przewlekłe choroby mieloproliferacyjne MPN CMPD 2. Zespoły mieloproliferacyjno-mielodysplastyczne MPD/MDS 3. Zespoły mielodysplastyczne MDS 4. Ostre białaczki szpikowe AML 5. Chłoniaki NHL, HL 6. Nowotwory mastocytów 7. Nowotwory histiocytów i kk prezentujących antygeny

14
zapadalność na 100 000 osób/ rok
ALL CLL/SLL szpiczak CML

15
Białaczki AML ALL = B/T-lymphoblastic leukemia (precursor lymphocytes)
CML (grupa MPN) Przewlekłe linii limfocyta – najczęściej – CLL
 Przewlekłe linii limfocyta – najczęściej – CLL.")
16
Diagnostyka hematopatologiczna
Dane kliniczne – dokładne!

17
AML? Neupogen! Dane kliniczne – dokładne!
Ca ventriculi post chemiotherapiam AML? Neupogen!

20
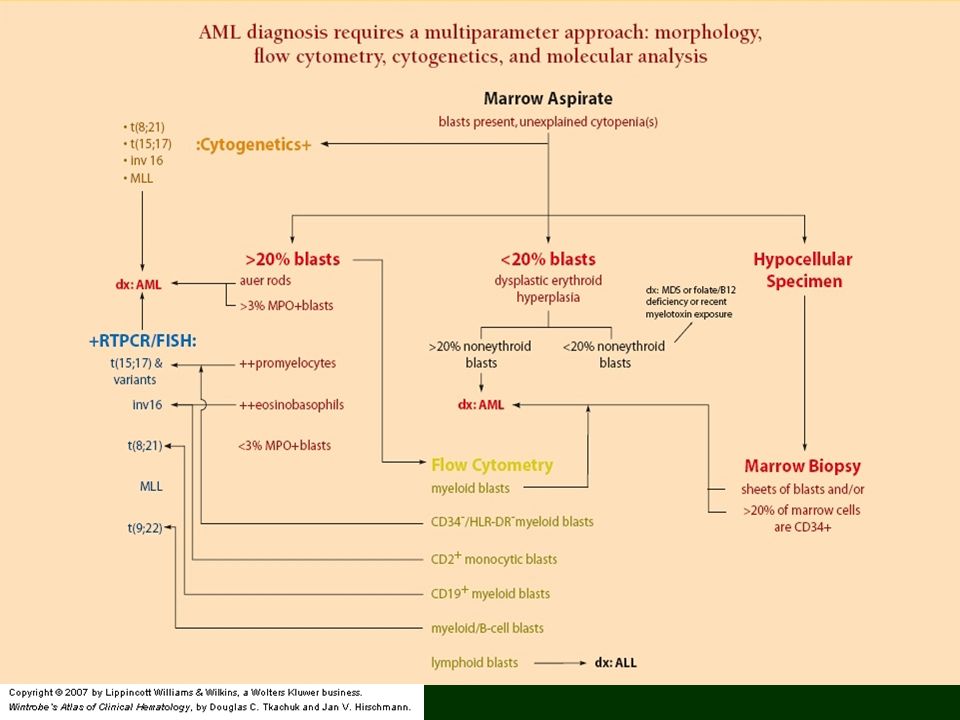
Ostra białaczka szpikowa - AML
Nowotworowa proliferacja hemopoetycznych komórek prekursorowych prowadząca do nadmiaru mieloblastów/innych niedojrzałych komórek szpikowych Czynniki ryzyka: większość pacjentów – żadne; Down’s syndrome, Bloom’s syndrome, Fanconi’s anemia, neurofibromatosis, ekspozycja na benzen, promieniowanie jonizujące, środki alkilujące, typ II inhibitorów topoizomerazy Objawy: męczliwość, osłabienie (bo anemia), gorączka, infekcje, skaza, nacieki tkanek (mielomonocytarna, monocytarna i monoblastyczna)(gingival hyperplasia, leukemia cutis); bóle mostka (ekspansja szpiku); zaburzenia neurologiczne
, gorączka, infekcje, skaza, nacieki tkanek (mielomonocytarna, monocytarna i monoblastyczna)(gingival hyperplasia, leukemia cutis); bóle mostka (ekspansja szpiku); zaburzenia neurologiczne.")
21
AML 50% WBC >10,000, >100,000 u 20%; aleukemic leukemia – brak blastów we krwi Prognostycznie korzystne: młody wiek, szybka odpowiedź na terapię, korzystna cytogenetyka Prognostycznie niekorzystne: <2 lub >60, wysoka leukocytoza przy diagnozie, MDS-related, mutacje FLT3 Leczenie: Chemioterapia - wyleczalność 10-30%, allogeniczny przeszczep szpiku %; 5-letnie przeżycie - 20% dorośli, 50% dzieci

22
AML - classification French-American-British (FAB) classification system , M0-M7 (30% blastów jako kryterium); WHO2008 Acute myeloid leukemias with recurrent genetic abnormalities: - AML with t(8;21)(q22;q22) (AML1/ETO), - AML with inv(16)(p13q22) or t(16;16)(p13;q22) (CBFβ/MYH11), - Acute promyelocytic leukemia (AML with t(15;17)(q22;q12) (PML/RARα) and variants, - AML with 11q23 (MLL) abnormalities Acute myeloid leukemia with multilineage dysplasia Acute myeloid leukemia and myelodysplastic syndrome, therapy related Acute myeloid leukemia not otherwise categorized: M0-M7 (M3 powyżej), Acute basophilic leukemia, Acute panmyelosis with myelofibrosis, Myeloid sarcoma Acute leukemia of ambiguous lineage: Undifferentiated acute leukemia, Bilineal acute leukemia, Biphenotypic acute leukemia
(q22;q22) (AML1/ETO), - AML with inv(16)(p13q22) or t(16;16)(p13;q22) (CBFβ/MYH11), - Acute promyelocytic leukemia (AML with t(15;17)(q22;q12) (PML/RARα) and variants, - AML with 11q23 (MLL) abnormalities. Acute myeloid leukemia with multilineage dysplasia. Acute myeloid leukemia and myelodysplastic syndrome, therapy related. Acute myeloid leukemia not otherwise categorized: M0-M7 (M3 powyżej), Acute basophilic leukemia, Acute panmyelosis with myelofibrosis, Myeloid sarcoma. Acute leukemia of ambiguous lineage: Undifferentiated acute leukemia, Bilineal acute leukemia, Biphenotypic acute leukemia.")
23
AML not otherwise categorized
Najczęściej: M2, M4, M1 Bardzo rzadkie M6, M7; M7 częsta - Down’s syndrome Acute monoblastic i acute monocytic leukemia (M5): 10% AML; częste zaburzenia krzępnięcia (m.in. DIC), organomegalia, limfadenopatia, gingival hyperplasia, zajęcie CNS i innych tkanek
: 10% AML; częste zaburzenia krzępnięcia (m.in. DIC), organomegalia, limfadenopatia, gingival hyperplasia, zajęcie CNS i innych tkanek.")
24
Acute promyelocytic leukemia (APL) with t(15;17)(q22;q12) (AML M3)
Hipergranularna (częściej) lub mikrogranularna 8% AML, 15% AML u dorosłych Najczęściej lat Niska WBC; zwykle DIC, organomegalia bardzo rzadko; All trans retinoic acid (ATRA) – różnicowanie promielocytów do neutrofili; przeżycie – świetne, przy kontroli DIC; Microgranular variant – zwykle wysoka wartość WBC
 lub mikrogranularna. 8% AML, 15% AML u dorosłych. Najczęściej lat. Niska WBC; zwykle DIC, organomegalia bardzo rzadko; All trans retinoic acid (ATRA) – różnicowanie promielocytów do neutrofili; przeżycie – świetne, przy kontroli DIC; Microgranular variant – zwykle wysoka wartość WBC.")
25
Myeloid sarcoma (extramedullary myeloid tumor, granulocytic sarcoma, chloroma)
Pozaszpikowy naciek z niedojrzałych komórek szpikowych 2-8% AML; najczęściej AML M4 lub M5, CML, PMF, HES, PV; prognoza jak dla odpowiedniej AML Rzadko – izolowany, bez współistniejącej choroby szpiku Węzły, podokostnowo, skóra, oczodół, kanał kręgowy, śródpiersie, jądra, jajniki

26
Acute bilineal leukemia Acute biphenotypic leukemia
Acute bilineal i biphenotypic leukemias – poniżej 4% AML; zwykle – złe rokowanie Bilineal – dwie odrębne populacje blastów, szpikowa i limfoidalna lub – rzadziej - B i T Biphenotypic – jedna populacja z ekspresją „mieszanki” liniowych markerów, zwykle – myeloid i B lub T-komórkowych

27
Myelodysplastic syndrome (MDS)
Klonalny rozrost komórek hemopoezy; nieefektywna hemopoeza – cytopenia (najczęściej – anemia) 25-45% - (AML) Średni wiek 65 lat; każdy wiek, ale rzadko poniżej 50rż. 50% początkowo – bez objawów
 25-45% - (AML) Średni wiek 65 lat; każdy wiek, ale rzadko poniżej 50rż. 50% początkowo – bez objawów.")
28
MDS Refractory anemia (RA, RARS)
Refractory cytopenia with unilineage dysplasia (RCUD) Refractory cytopenia with multilineage dysplasia (RCMD) RA with Excess Blasts (RAEB): type 1 has 5-9% blasts in blood/marrow, type 2 has 10-19% blasts in blood/marrow 5q- syndrome Therapy related MDS MDS, unclassified
 Refractory cytopenia with multilineage dysplasia (RCMD) RA with Excess Blasts (RAEB): type 1 has 5-9% blasts in blood/marrow, type 2 has 10-19% blasts in blood/marrow. 5q- syndrome. Therapy related MDS. MDS, unclassified.")
29
Zwykle anemia makrocytarna, niska/normalna retikulocytoza, różnie – neutropenia, trombocytopenia
Średnie przeżycia: RA -10 lat, RARS - 7 lat; RCMD - 3 lata; RAEB – 1 rok; therapy related MDS - 5 miesięcy; Śmierć – AML, krwawienia/infekcje

30
Myeloproliferative neoplasms (MPN) WHO 2008 = CMPD (WHO 2001)
Chronic myelogenous leukemia Polycythemia vera Essential thrombocythemia Primary myelofibrosis Chronic neutrophilic leukemia Chronic eosinophilic leukemia, not otherwise categorized; Hypereosinophilic syndrome Myeloproliferative neoplasms with rearrangement of PDGR alfa, beta, FGFR Mast cell disease MPNs, unclassifiable

31
MPN Efektywna klonalna mieloproliferacja, związana z różnymi mutacjami genów kinazy tyrozynowej (bcr-abl) lub pokrewnych białek – nieprawidłowa transdukcja sygnałów komórkowych Początkowo – wysoka komórkowość szpiku – wzrost hemopoezy, wzrost liczby krwinek, pozaszpikowa hemopoeza, potem – „spent phase” – włóknienie szpiku, cytopenia CML częsta progresja do AML; inne - rzadko Znaczące morfologiczne podobieństwo między jednostkami, a także między nimi i reaktywną hemopoezą
 lub pokrewnych białek – nieprawidłowa transdukcja sygnałów komórkowych. Początkowo – wysoka komórkowość szpiku – wzrost hemopoezy, wzrost liczby krwinek, pozaszpikowa hemopoeza, potem – „spent phase – włóknienie szpiku, cytopenia. CML częsta progresja do AML; inne - rzadko. Znaczące morfologiczne podobieństwo między jednostkami, a także między nimi i reaktywną hemopoezą.")
32
MPN Molecular CML - t(9;22)(q34;q11)-Philadelphia chromosome i/lub traskrypt fuzyjny genów ABL (9q34) i BCR (22q11) jest wymagany do diagnozy; JAK2 V617F mutation (gen Janus kinazy 2) – w większości nie-CML MPN (głównie - PV), powoduje ciągłą aktywność kinazy tyrozynowej; c-Mpl mutation (thrombopoietin receptor) – TE, PMF
 – w większości nie-CML MPN (głównie - PV), powoduje ciągłą aktywność kinazy tyrozynowej; c-Mpl mutation (thrombopoietin receptor) – TE, PMF.")
33
Chronic myelogenous leukemia (CML)
Najczęstszy MPN Z pluripotencjalnych komórek pnia, które mogą różnicować się do linii granulocyta lub limfocyta Powolny początek - anemia, osłabienie, utrata wagi Hepatosplenomegalia, może być limfadenopatia; zawały śledziony Bez leczenia: chronic phase 3-4 lata; 50% - accelerated phase, potem blast transformation zwykle 3 lata od początku (średnie przeżycie – bez leczenia – 3 lata); 70% AML, 30% ALL 100,000 WBC, z przesunięciem w lewo szeregu granulocyta, bazofilia, niska FAG, 50% - trombocytoza
; 70% AML, 30% ALL. 100,000 WBC, z przesunięciem w lewo szeregu granulocyta, bazofilia, niska FAG, 50% - trombocytoza.")
34
Polycythemia vera Klonalny rozrost nowotworowy multipotencjalnych komórek pnia Średnio 60 lat, rzadko u dzieci Zwykle niewielka hepatosplenomegalia Zaczerwienienie twarzy, sinica, nadciśnienie, zawroty, bóle głowy, dolegliwości brzuszne, świąd/owrzodzenia skóry – histamina z bazofilii; wtórna dna 25% powikłania zakrzepowe (udar, zawał serca, zakrzepica żył kończyn dolnych, Budd-Chiari) Prognoza: faza schyłkowa - 15% po 10 years, AML - 2% przy upustach; 15% przy chemioterapii
 Prognoza: faza schyłkowa - 15% po 10 years, AML - 2% przy upustach; 15% przy chemioterapii.")
35
Essential thrombocythemia
Średni lat, każdy wiek; 2/3 K Zwykle tylko płytki; >450,000 Zakrzepica i krwawienia Indolentna, przeżycie - norma

36
Primary myelofibrosis, PMF (2008, wcześniej – CIMF)
Średnio 60 years, rzadko u dzieci Klonalny defekt komórek pnia, zachowane dojrzewanie, progresywne włóknienie szpiku, pozaszpikowa hemopoeza; masywna splenomegalia (nawet 4kg) anemia, inne cytopenie, objawy B, dna, infekcje, zakrzepica, krwawienia Włóknienie – z nowotworowych megakariocytów - PDGF, bFGF, TGF beta i inne cytokiny 5% - AML Przeżycie lat
 anemia, inne cytopenie, objawy B, dna, infekcje, zakrzepica, krwawienia. Włóknienie – z nowotworowych megakariocytów - PDGF, bFGF, TGF beta i inne cytokiny. 5% - AML. Przeżycie 3.5-5lat.")
37
patologia grasicy

38
myasthenia gravis 65% - rozrost grasicy 25% - „normalna” grasica
10% - thymoma ryzyko grasiczaka – mężczyźni z pierwszymi objawami MG po 50r.ż. u 30-45% pacjentów z grasiczakiem – jednocześnie lub miesiące/lata po usunięciu leczenie usunięcie grasicy (nawet, gdy nie ma grasiczaka)
")
39
torbiele grasicy: -jednokomorowe (zwykle wrodzone) -wielokomorowe (zwykle nabyte) do 18cm górno-przednie śródpiersie

40
grasiczaki najczęstsze pierwotne nowotwory przedniego śródpiersia
rzadko – ektopowe – tylne śródpiersie, wnęka płuca, szyja, opłucna, tarczyca 80% otorebkowane, reszta – naciekanie wszystkie mogą być inwazyjne

41
klasyfikacja WHO A: nabłonkowy, wrzecionowatokomórkowy, rdzenny; monomorficzne komórki (wrzecionowate/owalne) bez atypii, mało/brak limfocytów; dobre rokowanie AB: mieszany; obszary jak A + obszary z liczniejszymi limfocytami B: przypomina grasicę płodu/niemowlaka B1 bogaty w limfocyty B2 korowy B3 niewielka atypia, mało limfocytów C: thymic carcinoma
 bez atypii, mało/brak limfocytów; dobre rokowanie. AB: mieszany; obszary jak A + obszary z liczniejszymi limfocytami. B: przypomina grasicę płodu/niemowlaka. B1 bogaty w limfocyty. B2 korowy. B3 niewielka atypia, mało limfocytów. C: thymic carcinoma.")
42
Inne nowotwory grasiy rak chłoniak potworniak

43
MAS (macrophage activation syndrome)
Kliniczno-patologiczna jednostka występująca w przebiegu różnych zespołów hemofagocytarnych: -pierwotna hemofagocytarna limfohistiocytoza (HLH) -wtórne zespoły hemofagocytarne (wtórna HLH)
 -wtórne zespoły hemofagocytarne (wtórna HLH)")
44
MAS – rzadka, ale często fatalna choroba; 15-60% śmiertelność (przy leczeniu)
Zwykle „ostra”: gorączka, limfadenopatia, pancytopenia, dysfunkcja wątroby, hipertrójglicerydemia, hiperferrytynemia; rozwój koagulopatii i dysfunkcji CSN; Niekontrolowana aktywacja i proliferacja limfocytów T i nadmierna aktywacja makrofagów; Genetyczna – defekt „zabójczych” właściwości CTL lub NK (20-40% - mutacja perforyny)
")
45
Wtórne HSs Infekcje Nowotwory
Choroby immunologiczne (SLE, juvenile RA) Wzrost poziomu INFgamma, TNFalfa, IL1, IL6
 Wzrost poziomu INFgamma, TNFalfa, IL1, IL6.")
46
Familial HLH (Farquhar disease)
Acquired HLH Exogenous agents (infectious organisms, toxins) Infection-associated hemophagocytic syndrome (IAHS) Endogenous products (tissue damage, metabolic products) Rheumatic diseases macrophage activation syndrome (MAS) Malignant diseases Genetic HLH Familial HLH (Farquhar disease) Known gene defects (perforin, munc 13-4, syntaxin 11) Unknown gene defects Immune deficiency syndromes Chédiak-Higashi syndrome (CHS) Griscelli syndrome (GS) X-linked lymphoproliferative syndrome (XLP) Hematology 2005
 Infection-associated hemophagocytic syndrome (IAHS) Endogenous products (tissue damage, metabolic products) Rheumatic diseases macrophage activation syndrome (MAS) Malignant diseases. Genetic HLH Familial HLH (Farquhar disease) Known gene defects (perforin, munc 13-4, syntaxin 11) Unknown gene defects Immune deficiency syndromes Chédiak-Higashi syndrome (CHS) Griscelli syndrome (GS) X-linked lymphoproliferative syndrome (XLP) Hematology")
47
revised criteria of the Histiocyte Society for the diagnosis of HLH
Familial disease/known genetic defect Clinical and laboratory criteria (5/8 criteria) Fever Splenomegaly Cytopenia 2 cell lines Hemoglobin < 90 g/L (below 4 weeks < 120 g/L) Neutrophils < 1 x 109/L Hypertriglyceridemia and/or hypofibrinogenemia Ferritin µg/L sCD U/mL Decreased or absent NK-cell activity Hemophagocytosis in bone marrow, CSF or lymph nodes Hematology 2005
 Fever. Splenomegaly. Cytopenia 2 cell lines. Hemoglobin < 90 g/L (below 4 weeks < 120 g/L) Neutrophils < 1 x 109/L. Hypertriglyceridemia and/or hypofibrinogenemia. Ferritin 500 µg/L. sCD U/mL. Decreased or absent NK-cell activity. Hemophagocytosis in bone marrow, CSF or lymph nodes. Hematology")
48
Mężczyzna l. 22 XII 2007/I 2008 węzeł chłonny – uogólnione powiększenie węzłów chłonnych - węzeł (o maks. wymiarze 22mm)
 .")
49
Trepan V 2008 Stan ciężki; pancytopenia (brak danych liczbowych), hepatosplenomegalia, obrzęk mózgu, uogólniona limfadenomegalia, gorączka Hospitalizowany w Klinice Ch. Zakaźnych (na respiratorze); wykluczono infekcje
; wykluczono infekcje.")
Podobne prezentacje












 i WTÓRNE (NABYTE) NIEDOBORY ODPORNOŚCI>")


 Ostre Przewlekłe OSTRE:>")



