Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Hematopatologia

2
Układ czerwonokrwinkowy
Anemia Z utraty krwi: ostra, przewlekła Spadek produkcji: defekt dojrzewania (megaloblastyczna, niedobór żelaza, MDS), zaburzenia proliferacji (defekt komórek pnia, wyparcie hemopoezy, anemia chorób przewlekłych) Hemolityczna: zaburzenia struktury krwinek – hemoglobinopatie (thalassemia, anemia sierpowata), nieprawidłowości błony (wrodzona sferocytoza, eliptocytoza), nieprawidłowości enzymatyczne (niedobór dehydrogenzy glukozo-6-fosforanowej), PNH (defekty cząstek łączących się z fosfatydyloinozytolem – genetyczny defekt nabyty) Czynniki zewnętrzne – AIHA (ciepłe i zimne p/ciała), mikroangiopatyczna (m.in. HUS), pasożytnicze (malaria, babesioza) Erytrocytoza
, zaburzenia proliferacji (defekt komórek pnia, wyparcie hemopoezy, anemia chorób przewlekłych) Hemolityczna: zaburzenia struktury krwinek – hemoglobinopatie (thalassemia, anemia sierpowata), nieprawidłowości błony (wrodzona sferocytoza, eliptocytoza), nieprawidłowości enzymatyczne (niedobór dehydrogenzy glukozo-6-fosforanowej), PNH (defekty cząstek łączących się z fosfatydyloinozytolem – genetyczny defekt nabyty) Czynniki zewnętrzne – AIHA (ciepłe i zimne p/ciała), mikroangiopatyczna (m.in. HUS), pasożytnicze (malaria, babesioza) Erytrocytoza.")
3
Anemia = spadek Hb Normo-/mikro-/makrocytarna
Normo-/hipo-/hiperchromiczna

4
Anemia makrocytarna Noworodki, alkoholizm, niedobór kw. foliowego, B12, choroby wątroby, niedoczynność tarczycy, MDS, AA, HCL, ostre białaczki, leki: cyklofosfamid, metotreksat, antywirusowe, hipoglikemiczne (metformina), p/drgawkowe (fenytoina), p/zapalne (sulfasalazyna) Megaloblastyczna (B12) MCV zwykle powyżej 115; pancytopenia; 10 dni od leczenia – wzrost Hb; do normy – 8 tygodni; możliwe zaburzenia neurologiczne; wzrost ryzyka raka żołądka i NET żołądka (anemia złośliwa)
, p/drgawkowe (fenytoina), p/zapalne (sulfasalazyna) Megaloblastyczna (B12) MCV zwykle powyżej 115; pancytopenia; 10 dni od leczenia – wzrost Hb; do normy – 8 tygodni; możliwe zaburzenia neurologiczne; wzrost ryzyka raka żołądka i NET żołądka (anemia złośliwa)")
5
Anemia mikrocytarna Niedobór Fe, thalassemia, choroby przewlekłe, anemia sideroblastyczna, niedobór Cu, zatrucie Zn Z niedoboru Fe – I miejsce na świecie – najczęściej: kobiety (wiek rozrodczy i w ciąży), wcześniaki; słaba korelacja między objawami a poziomem Hb (inne enzymy z Fe); wczesny możliwy objaw – pica – patologiczne „smaki” (na niejadalne); wzrost Hb po 2 tygodniach, norma po 2 miesiącach; u starszych – kał na krew utajoną, gastro-/kolonoskopia
, wcześniaki; słaba korelacja między objawami a poziomem Hb (inne enzymy z Fe); wczesny możliwy objaw – pica – patologiczne „smaki (na niejadalne); wzrost Hb po 2 tygodniach, norma po 2 miesiącach; u starszych – kał na krew utajoną, gastro-/kolonoskopia.")
6
Anemia sierpowata Przewlekła anemia hemolityczna + okluzja mikrokrążenia – polimeryzacja odtlenowanej Hb S; Większa wrażliwość na infekcję parwowirusem B19 – kryza aplastyczna; Zawały śledziony, martwice kości, ryzyko udaru, dysfunkcja nerek, żółtaczka, kamica żółciowa; rak rdzeniasty nerki

7
Thalassemia Hipochromiczna, mikrocytarna anemia; spadek produkcji łańcuchów globiny alfa lub beta Deformacje szkieletu, hepatosplenomegalia, wtórna hemochromatoza

8
AIHA Ciepłe – IgG (37oC); częściej; pierwotna (idiopatyczna), wtórna – choroby autoimmunologiczne, limfoproliferacje, leki; splenomegalia (eliminacja w śledzionie); każdy wiek, K:M = 2:1; Zimne – IgM aglutyniny; pierwotna – przewlekła, głównie kobiety w starszym wieku; wtórna – głównie limfoproliferacje – m.in. makroglobulinemia Waldenstroema; infekcje – Mycoplasma pneumoniae, mononukleoza – zwykle samoistnie zanika w ciągu kilka tygodni; rzadko organomegalia
; częściej; pierwotna (idiopatyczna), wtórna – choroby autoimmunologiczne, limfoproliferacje, leki; splenomegalia (eliminacja w śledzionie); każdy wiek, K:M = 2:1; Zimne – IgM aglutyniny; pierwotna – przewlekła, głównie kobiety w starszym wieku; wtórna – głównie limfoproliferacje – m.in. makroglobulinemia Waldenstroema; infekcje – Mycoplasma pneumoniae, mononukleoza – zwykle samoistnie zanika w ciągu kilka tygodni; rzadko organomegalia.")
9
Erytrocytoza Spadek objętości osocza (względna)
Wzrost masy erytrocytów – policytemia: Pierwotna: rodzinna, PV Wtórna: choroby serca/płuc, duża wysokość n.p.m.; obniżone uwalnianie tlenu z Hb (wrodzone warianty o wysokim powinowactwie do tlenu, karboksy-Hb u palaczy); ektopowa produkcja EPO
; ektopowa produkcja EPO.")
10
Leukocytoza/leukopenia
Leukocytoza reaktywna – limfocytarna przy infekcjach wirusowych; reakcje białaczkowe; poliklonalna limfocytoza u palaczek (HLA-DR7); Leukopenia – polekowa, przy chorobach autoimmunologicznych
; Leukopenia – polekowa, przy chorobach autoimmunologicznych.")
11
Płytki Trombocytoza (możliwe – powikłania krwotoczne, zakrzepowe lub – bezobjawowo): Reaktywna – niedobór żelaza, nowotwory, zapalenia, infekcje, po splenektomii, po odstawieniu leków mielosupresyjnych Nowotworowe – TE, PV, PMF, CML (może być w pewnych postaciach AML – inv3) Trombocytopenia – ITP, DIC, indukowana heparyną, zakrzepowe (TTP, HUS) Skazy osoczowe, naczyniowe (główne cechy; hemofilia A i B)
 Trombocytopenia – ITP, DIC, indukowana heparyną, zakrzepowe (TTP, HUS) Skazy osoczowe, naczyniowe (główne cechy; hemofilia A i B)")
12
Nowotwory układu krwiotwórczego
1. Przewlekłe choroby mieloproliferacyjne=nowotwory mieloproliferacyjne CMPD (MPN) 2. choroby (nowotwory) mielodysplastyczno-mieloproliferacyjne MDS/MPN 3. Zespoły mielodysplastyczne MDS 4. Ostre białaczki szpikowe AML 5. Chłoniaki NHL, HL 6. Nowotwory mastocytów 7. Nowotwory histiocytów i kk prezentujących antygeny
 2. choroby (nowotwory) mielodysplastyczno-mieloproliferacyjne MDS/MPN. 3. Zespoły mielodysplastyczne MDS. 4. Ostre białaczki szpikowe AML. 5. Chłoniaki NHL, HL. 6. Nowotwory mastocytów. 7. Nowotwory histiocytów i kk prezentujących antygeny.")
13
zapadalność na 100 000 osób/ rok
ALL CLL/SLL szpiczak CML

14
Białaczki AML ALL = B/T lymphoblastic leukemia (precursor lymphocytes)
CML (grupa MPN) Przewlekłe linii limfocyta – najczęściej – CLL
 Przewlekłe linii limfocyta – najczęściej – CLL.")
15
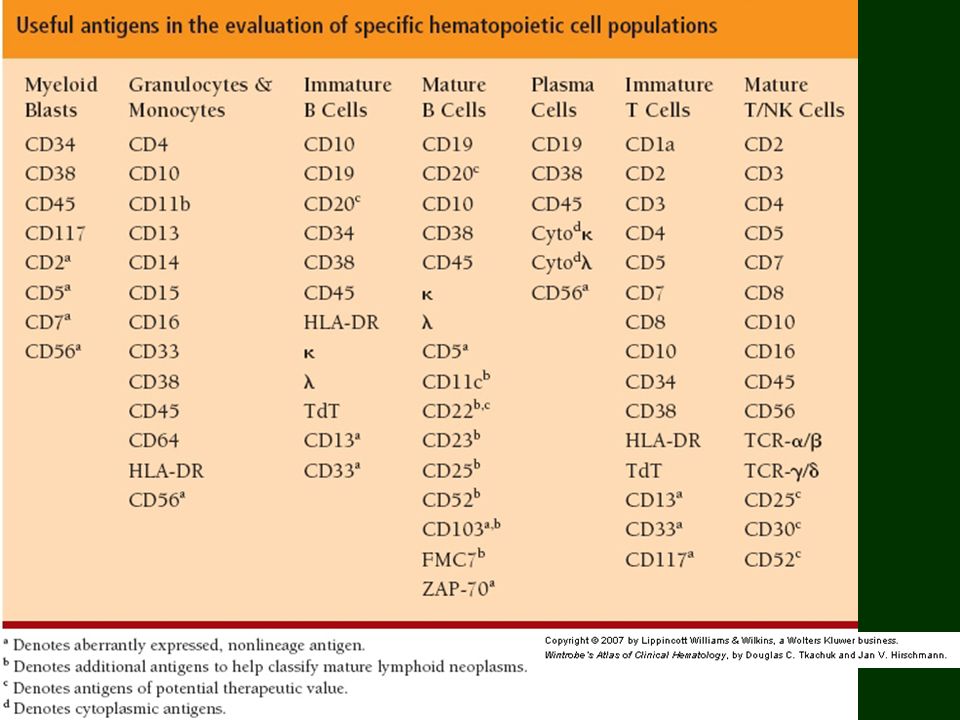
Diagnostyka hematopatologiczna
Dane kliniczne – dokładne! Materiał – aspiraty – rozmazy, FC; materiał histopatologiczny: węzeł, trepanobiopsja, śledziona, wycinki; Metody – histopatologia (+IHC), cytologia, FC, molekularne
, cytologia, FC, molekularne.")
18
BAC w porównaniu z pełną oceną histologiczną:
75%: prawidłowe rozróżnienie chłoniak / odczyn 49% % prawidłowo sklasyfikowanych chłoniaków Diagn Cytopathol 1999, 22:336-41 (Univ. Utah, Univ. Pennsylvania, Univ. Washington)
")
19
wywiad objawy "B" inne systemowe organomegalia morfologia historia leki nowotwory

20
Zagadnienie biopsji śledziony
splenomegalia norma: 13 cm w osi długiej < 250 g (zwykle ok. 150 g) niewielka splenomegalia: palpacja daje wyniki negatywne w 30%-40% przyczyny: 1. rozrost/ przerost z powodu nadczynności rozpad krwinek, zjawiska odpornościowe, autoimmunizacja 2. przekrwienie bierne 3. choroba naciekowa npl, choroba spichrzeniowa, amyloidoza
 niewielka splenomegalia: palpacja daje wyniki negatywne w 30%-40% przyczyny: 1. rozrost/ przerost z powodu nadczynności. rozpad krwinek, zjawiska odpornościowe, autoimmunizacja. 2. przekrwienie bierne. 3. choroba naciekowa. npl, choroba spichrzeniowa, amyloidoza.")
21
MASYWNA splenomegalia > 1000 g lub > 8 cm poniżej łuku żebrowego
w praktyce lista ograniczona do: przewlekłe choroby mieloproliferacyjne*: CML PMF PV chłoniaki nieziarnicze, w tym zwłaszcza*: białaczka włochatokomórkowa przewlekła białaczka limfocytarna choroba Gauchera* sarkoidoza autoimmunizacyjna anemia hemolityczna rozlana naczyniakowatość śledziony

22
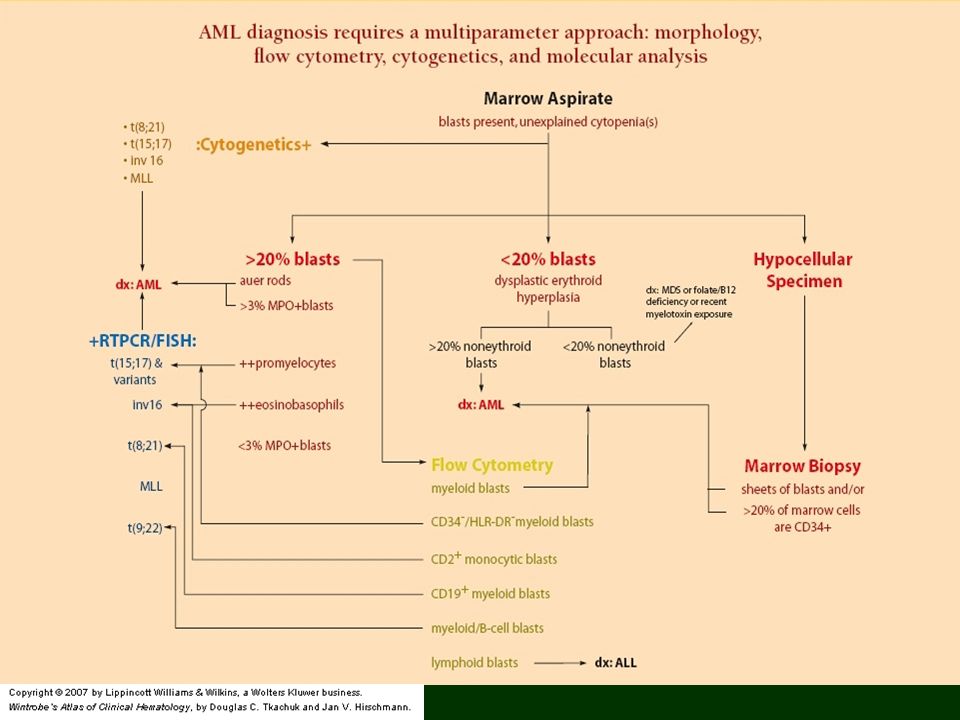
Ostra białaczka szpikowa - AML
Nowotworowa proliferacja hemopoetycznych komórek prekursorowych prowadząca do nadmiaru mieloblastów/innych niedojrzałych komórek szpikowych Czynniki ryzyka: większość pacjentów – żadne; Down’s syndrome, Bloom’s syndrome, Fanconi’s anemia, neurofibromatosis, ekspozycja na benzen, promieniowanie jonizujące, środki alkilujące, typ II inhibitorów topoizomerazy Objawy: męczliwość, osłabienie (bo anemia), gorączka, infekcje, skaza, nacieki tkanek (mielomonocytarna, monocytarna i monoblastyczna)(gingival hyperplasia, leukemia cutis); bóle mostka (ekspansja szpiku); zaburzenia neurologiczne
, gorączka, infekcje, skaza, nacieki tkanek (mielomonocytarna, monocytarna i monoblastyczna)(gingival hyperplasia, leukemia cutis); bóle mostka (ekspansja szpiku); zaburzenia neurologiczne.")
23
AML 50% WBC >10,000, >100,000 u 20%; aleukemic leukemia – brak blastów we krwi Prognostycznie korzystne: młody wiek, szybka odpowiedź na terapię, korzystna cytogenetyka Prognostycznie niekorzystne: <2 lub >60, wysoka leukocytoza przy diagnozie, MDS-related, mutacje FLT3 Leczenie: Chemioterapia - wyleczalność 10-30%, allogeniczny przeszczep szpiku %; 5-letnie przeżycie - 20% dorośli, 50% dzieci

24
AML - classification French-American-British (FAB) classification system , M0-M7 (30% blastów jako kryterium); WHO2008 Acute myeloid leukemias with recurrent genetic abnormalities: Acute promyelocytic leukemia (AML with t(15;17)(q22;q12) (PML/RARα) Acute myeloid leukemia with multilineage dysplasia Acute myeloid leukemia and myelodysplastic syndrome, therapy related Acute myeloid leukemia not otherwise categorized: M0-M7 (M3 powyżej), Acute basophilic leukemia, Acute panmyelosis with myelofibrosis, Myeloid sarcoma Acute leukemia of ambiguous lineage: Undifferentiated acute leukemia, Bilineal acute leukemia, Biphenotypic acute leukemia
(q22;q12) (PML/RARα) Acute myeloid leukemia with multilineage dysplasia. Acute myeloid leukemia and myelodysplastic syndrome, therapy related. Acute myeloid leukemia not otherwise categorized: M0-M7 (M3 powyżej), Acute basophilic leukemia, Acute panmyelosis with myelofibrosis, Myeloid sarcoma. Acute leukemia of ambiguous lineage: Undifferentiated acute leukemia, Bilineal acute leukemia, Biphenotypic acute leukemia.")
25
AML not otherwise categorized
Najczęściej: M2, M4, M1 Bardzo rzadkie M6, M7; M7 częsta - Down’s syndrome Acute monoblastic i acute monocytic leukemia (M5): 10% AML; częste zaburzenia krzępnięcia (m.in. DIC), organomegalia, limfadenopatia, gingival hyperplasia, zajęcie CNS i innych tkanek
: 10% AML; częste zaburzenia krzępnięcia (m.in. DIC), organomegalia, limfadenopatia, gingival hyperplasia, zajęcie CNS i innych tkanek.")
26
Acute promyelocytic leukemia (APL) with t(15;17)(q22;q12) (AML M3)
Hipergranularna (częściej) lub mikrogranularna 8% AML, 15% AML u dorosłych Najczęściej lat Niska WBC; zwykle DIC, organomegalia bardzo rzadko; All trans retinoic acid (ATRA) – różnicowanie promielocytów do neutrofili; przeżycie – świetne, przy kontroli DIC; Microgranular variant – zwykle wysoka wartość WBC
 lub mikrogranularna. 8% AML, 15% AML u dorosłych. Najczęściej lat. Niska WBC; zwykle DIC, organomegalia bardzo rzadko; All trans retinoic acid (ATRA) – różnicowanie promielocytów do neutrofili; przeżycie – świetne, przy kontroli DIC; Microgranular variant – zwykle wysoka wartość WBC.")
27
Myeloid sarcoma (extramedullary myeloid tumor, granulocytic sarcoma, chloroma)
Pozaszpikowy naciek z niedojrzałych komórek szpikowych 2-8% AML; najczęściej AML M4 lub M5, CML, PMF, HES, PV; prognoza jak dla odpowiedniej AML Rzadko – izolowany, bez współistniejącej choroby szpiku Węzły, podokostnowo, skóra, oczodół, kanał kręgowy, śródpiersie, jądra, jajniki

28
ALL - classification WHO classification of Acute Lymphoblastic Leukemia B-lymphoblastic leukemia / lymphoblastic lymphoma, NOS - ALL with t(9;22)(q34;q11) (BCR-ABL-Philadelphia chromosome) - ALL with t(v;11q23) (MLL rearranged) - ALL with t(1;19)(q23;p13) (PBX-E2A) - ALL with t(12;21)(p13;q22) (TEL-AML1) - Hyperdiploid >50 - Hypodiploid T-lymphoblastic leukemia / lymphoma
(q34;q11) (BCR-ABL-Philadelphia chromosome) - ALL with t(v;11q23) (MLL rearranged) - ALL with t(1;19)(q23;p13) (PBX-E2A) - ALL with t(12;21)(p13;q22) (TEL-AML1) - Hyperdiploid >50. - Hypodiploid. T-lymphoblastic leukemia / lymphoma.")
29
B lymphoblastic leukemia (B-ALL) / lymphoblastic lymphoma (B-LBL)
Zwykle dzieci B ALL – pancytopenia, nagły początek objawów, bóle kostne, hepatosplenomegalia, objawy CSN (zajęcie opon), powiększenie jąder B-lymphoblastic lymphoma – zwykle – guzki skórne, zajęcie kości lub węzłów, BEZ zajęcia szpiku
, powiększenie jąder. B-lymphoblastic lymphoma – zwykle – guzki skórne, zajęcie kości lub węzłów, BEZ zajęcia szpiku.")
30
Najlepsza prognoza: 2-10 lat, early preB phenotype, hyperdiploidia, t(12;21)
Zła prognoza: < 2 lat, >10 lat, t(9;22), CD10- lub ekspresja antygenów mieloidalnych (np. MPO) B ALL with t(9;22)(q34;q11) - z fuzyjnym transkryptem bcr-abl (Philadelphia chromosome); 30% dorosłych ALL, 4% dzieci - ale - 80% niemowląt; poza niemowlętami – zwykle w starszym wieku, wyższa WBC, częściej organomegalia i zajęcie CSN, złe rokowanie
, CD10- lub ekspresja antygenów mieloidalnych (np. MPO) B ALL with t(9;22)(q34;q11) - z fuzyjnym transkryptem bcr-abl (Philadelphia chromosome); 30% dorosłych ALL, 4% dzieci - ale - 80% niemowląt; poza niemowlętami – zwykle w starszym wieku, wyższa WBC, częściej organomegalia i zajęcie CSN, złe rokowanie.")
31
T lymphoblastic leukemia / lymphoma (T-ALL/LBL)
Nastolatki, młodzi mężczyźni (starsi niż B-ALL/LBL) T ALL 15% dziecięcych i 20-25% dorosłych ALL T LBL % LBL, zwykle – guz śródpiersia bez/z minimalnym zajęciem szpiku Zajęcie CNS częściej niż B T-ALL 25% lub więcej komórek szpiku lub – nie ma „guza” Wyleczalność 60%; wcześniejsze wznowy i gorsze rokowanie niż B ALL
 T ALL 15% dziecięcych i 20-25% dorosłych ALL T LBL % LBL, zwykle – guz śródpiersia bez/z minimalnym zajęciem szpiku. Zajęcie CNS częściej niż B. T-ALL 25% lub więcej komórek szpiku lub – nie ma „guza Wyleczalność 60%; wcześniejsze wznowy i gorsze rokowanie niż B ALL.")
32
Acute bilineal leukemia Acute biphenotypic leukemia
Acute bilineal i biphenotypic leukemias – poniżej 4% AML; zwykle – złe rokowanie Bilineal – dwie odrębne populacje blastów, szpikowa i limfoidalna lub – rzadziej - B i T Biphenotypic – jedna populacja z ekspresją „mieszanki” liniowych markerów, zwykle – myeloid i B lub T-komórkowych

33
Myelodysplastic syndrome (MDS)
Klonalny rozrost komórek hemopoezy; nieefektywna hemopoeza – cytopenia (najczęściej – anemia) 25-45% - (AML) Średni wiek 65 lat; każdy, ale rzadko poniżej 50rż. 50% początkowo – bez objawów
 25-45% - (AML) Średni wiek 65 lat; każdy, ale rzadko poniżej 50rż. 50% początkowo – bez objawów.")
34
MDS Refractory anemia (RA, RARS)
Refractory cytopenia with unilineage dysplasia (RCUD) Refractory cytopenia with multilineage dysplasia (RCMD) RA with Excess Blasts (RAEB): type 1 has 5-9% blasts in blood/marrow, type 2 has 10-19% blasts in blood/marrow 5q- syndrome Therapy related MDS MDS, unclassified
 Refractory cytopenia with multilineage dysplasia (RCMD) RA with Excess Blasts (RAEB): type 1 has 5-9% blasts in blood/marrow, type 2 has 10-19% blasts in blood/marrow. 5q- syndrome. Therapy related MDS. MDS, unclassified.")
35
Zwykle anemia makrocytarna, niska/normalna retikulocytoza, różnie – neutropenia, trombocytopenia
Średnie przeżycia: RA -10 lat, RARS - 7 lat; RCMD - 3 lata; RAEB – 1 rok; therapy related MDS - 5 miesięcy; Śmierć – AML, krwawienia/infekcje

36
Myeloproliferative neoplasms (MPN) WHO 2008 = CMPD (WHO 2001)
Chronic myelogenous leukemia Polycythemia vera Essential thrombocythemia Primary myelofibrosis Chronic neutrophilic leukemia Chronic eosinophilic leukemia, not otherwise categorized; Hypereosinophilic syndrome Myeloproliferative neoplasms with rearrangement of PDGR alfa, beta, FGFR Mast cell disease MPNs, unclassifiable

37
MPN Efektywna klonalna mieloproliferacja, związana z różnymi mutacjami genów kinazy tyrozynowej (bcr-abl) lub pokrewnych białek – nieprawidłowa transdukcja sygnałów komórkowych Początkowo – wysoka komórkowość szpiku – wzrost hemopoezy, wzrost liczby krwinek, pozaszpikowa hemopoeza, potem – „spent phase” – włóknienie szpiku, cytopenia CML częsta progresja do AML; inne - rzadko Znaczące morfologiczne podobieństwo między jednostkami, a także między nimi i reaktywną hemopoezą
 lub pokrewnych białek – nieprawidłowa transdukcja sygnałów komórkowych. Początkowo – wysoka komórkowość szpiku – wzrost hemopoezy, wzrost liczby krwinek, pozaszpikowa hemopoeza, potem – „spent phase – włóknienie szpiku, cytopenia. CML częsta progresja do AML; inne - rzadko. Znaczące morfologiczne podobieństwo między jednostkami, a także między nimi i reaktywną hemopoezą.")
38
MPN Molecular CML - t(9;22)(q34;q11)-Philadelphia chromosome i/lub traskrypt fuzyjny genów ABL (9q34) i BCR (22q11) jest wymagalny do diagnozy; JAK2 V617F mutation (gen Janus kinazy 2) – w większości nie-CML MPN (głównie - PV), powoduje ciągłą aktywność kinazy tyrozynowej; c-Mpl mutation (thrombopoietin receptor) – TE, PMF
 – w większości nie-CML MPN (głównie - PV), powoduje ciągłą aktywność kinazy tyrozynowej; c-Mpl mutation (thrombopoietin receptor) – TE, PMF.")
39
Chronic myelogenous leukemia (CML)
Najczęstszy MPN Z pluripotencjalnych komórek pnia, które mogą różnicować się do linii granulocyta lub limfocyta Powolny początek - anemia, osłabienie, utrata wagi Hepatosplenomegalia, może być limfadenopatia; zawały śledziony Bez leczenia: chronic phase 3-4 lata; 50% - accelerated phase, potem blast transformation zwykle 3 lata od początku (średnie przeżycie – bez leczenia – 3 lata); 70% AML, 30% ALL 100,000 WBC, z przesunięciem w lewo szeregu granulocyta, bazofilia, niska FAG, 50% - trombocytoza
; 70% AML, 30% ALL. 100,000 WBC, z przesunięciem w lewo szeregu granulocyta, bazofilia, niska FAG, 50% - trombocytoza.")
40
Polycythemia vera Klonalny rozrost nowotworowy multipotencjalnych komórek pnia Średnio 60 lat, rzadko u dzieci Zwykle niewielka hepatosplenomegalia Zaczerwienienie twarzy, sinica, nadciśnienie, zawroty, bóle głowy, dolegliwości brzuszne, świąd/owrzodzenia skóry – histamina z bazofili; wtórna dna 25% powikłania zakrzepowe (udar, zawał serca, zakrzepica żył kończyn dolnych, Budd-Chiari) Prognoza: faza schyłkowa - 15% po 10 years, AML - 2% przy upustach; 15% przy chemioterapii
 Prognoza: faza schyłkowa - 15% po 10 years, AML - 2% przy upustach; 15% przy chemioterapii.")
41
Essential thrombocythemia
Średni lat, każdy wiek; 2/3 K Zwykle tylko płytki; >450,000 Zakrzepica i krwawienia Indolentna, przeżycie - norma

42
Primary myelofibrosis, PMF (2008, wcześniej – CIMF)
Średnio 60 lat, rzadko u dzieci Klonalny defekt komórek pnia, zachowane dojrzewanie, progresywne włóknienie szpiku, pozaszpikowa hemopoeza; masywna splenomegalia (nawet 4kg) anemia, inne cytopenie, objawy B, dna, infekcje, zakrzepica, krwawienia Włóknienie – z nowotworowych megakariocytów - PDGF, bFGF, TGF beta i inne cytokiny 5% - AML Przeżycie lat
 anemia, inne cytopenie, objawy B, dna, infekcje, zakrzepica, krwawienia. Włóknienie – z nowotworowych megakariocytów - PDGF, bFGF, TGF beta i inne cytokiny. 5% - AML. Przeżycie 3.5-5lat.")
43
patologia grasicy

44
myasthenia gravis 65% - rozrost grasicy 25% - „normalna” grasica
10% - thymoma ryzyko grasiczaka – mężczyźni z pierwszymi objawami MG po 50r.ż. u 30-45% pacjentów z grasiczakiem – jednocześnie lub miesiące/lata po usunięciu leczenie usunięcie grasicy (nawet, gdy nie ma grasiczaka)
")
45
torbiele grasicy: -jednokomorowe (zwykle wrodzone) -wielokomorowe (zwykle nabyte) do 18cm górno-przednie śródpiersie

46
grasiczaki najczęstsze pierwotne nowotwory przedniego śródpiersia
rzadko – ektopowe – tylne śródpiersie, wnęka płuca, szyja, opłucna, tarczyca 80% otorebkowane, reszta – naciekanie wszystkie mogą być inwazyjne

47
klasyfikacja WHO A: nabłonkowy, wrzecionowatokomórkowy, rdzenny; monomorficzne komórki (wrzecionowate/owalne) bez atypii, mało/brak limfocytów; dobre rokowanie AB: mieszany; obszary jak A + obszary z liczniejszymi limfocytami B: przypomina grasicę płodu/niemowlaka B1 bogaty w limfocyty B2 korowy B3 niewielka atypia, mało limfocytów C: thymic carcinoma
 bez atypii, mało/brak limfocytów; dobre rokowanie. AB: mieszany; obszary jak A + obszary z liczniejszymi limfocytami. B: przypomina grasicę płodu/niemowlaka. B1 bogaty w limfocyty. B2 korowy. B3 niewielka atypia, mało limfocytów. C: thymic carcinoma.")
Podobne prezentacje





 i WTÓRNE (NABYTE) NIEDOBORY ODPORNOŚCI>")


 Ostre Przewlekłe OSTRE:>")










