Pobierz prezentację
1
TRUDNOŚCI DIAGNOSTYCZNE U PACJENTÓW Z ZABURZENIAMI GOSPODARKI WAPNIOWO- FOSFORANOWEJ. MICHAŁ POPOW Katedra i Klinika Chorób Wewnętrznych i Endokrynologii Warszawskiego Uniwersytetu Medycznego Kierownik: prof. dr hab. med. Tomasz Bednarczuk XXI Zjazd Polskiego Towarzystwa Endokrynologicznego Katowice 2016

2
NADCZYNNOŚĆ PRZYTARCZYC Charakterystyczny wywiad lub przypadkowe wykrycie hiperkalcemii. Charakterystyczne odchylenia w badaniach biochemicznych: 1)Hiperkalcemia 2)Hipofosfatemia 3)Hiperkalciuria 4)Nieobniżony poziom PTH mimo hiperkalcemii
Hiperkalcemia 2)Hipofosfatemia 3)Hiperkalciuria 4)Nieobniżony poziom PTH mimo hiperkalcemii.")
3
GRUCZOLAK PRZYTARCZYC Gruczolak przytarczyc (adenoma) to monoklonalny rozrost wywodzący się z pojedynczej zmutowanej komórki. Mutacja najczęściej ma charakter nabyty. Wyjątek stanowi gruczolak u osób z mutacją genu parafibrominy (CDC73).
..")
4
JAKA JEST ETIOLOGIA NAJCZĘSTSZEJ POSTACI PIERWOTNEJ NADCZYNNOŚCI PRZYTARCZYC CZYLI POJEDYNCZEGO GRUCZOLAKA? MutacjaKomentarz VDR – receptora witaminy D (zmniejszająca jego wrażliwość na kalcytriol) Mutacja powoduje słabsze pobudzenie receptora witaminy w obrębie przytarczyc, co sprzyja proliferacji komórek gruczołu. Zaburzenia funkcji VDR rzadko spotykane są w gruczolakach, natomiast wiązane są z predyspozycją do przemiany złośliwej we wtórnej i trzeciorzędowej nadczynności przytarczyc. CaSR – mutacja receptora wapniowego (zmniejszająca jego wrażliwość na jony wapnia) Mutacja powoduje słabsze hamowanie aktywności wewnątrzydzielniczej komórek przytarczyc, co sprzyja ich namnażaniu i powiększaniu się. Zaburzenia funkcji CASR rzadko spotykane są w gruczolakach, natomiast podobnie jak w przypadku VDR wiązane są z predyspozycją do przemiany złośliwej we wtórnej i trzeciorzędowej nadczynności przytarczyc.
 Mutacja powoduje słabsze pobudzenie receptora witaminy w obrębie przytarczyc, co sprzyja proliferacji komórek gruczołu. Zaburzenia funkcji VDR rzadko spotykane są w gruczolakach, natomiast wiązane są z predyspozycją do przemiany złośliwej we wtórnej i trzeciorzędowej nadczynności przytarczyc. CaSR – mutacja receptora wapniowego (zmniejszająca jego wrażliwość na jony wapnia) Mutacja powoduje słabsze hamowanie aktywności wewnątrzydzielniczej komórek przytarczyc, co sprzyja ich namnażaniu i powiększaniu się. Zaburzenia funkcji CASR rzadko spotykane są w gruczolakach, natomiast podobnie jak w przypadku VDR wiązane są z predyspozycją do przemiany złośliwej we wtórnej i trzeciorzędowej nadczynności przytarczyc..")
5
JAKA JEST ETIOLOGIA NAJCZĘSTSZEJ POSTACI PIERWOTNEJ NADCZYNNOŚCI PRZYTARCZYC CZYLI POJEDYNCZEGO GRUCZOLAKA? PRAD1 – (szacowany udział w rozwoju gruczolaka przytarczyc: 20-40%) mutacja genu (pełniącego funkcję onkogenu) kodującego cyklinę D1. Prowadzi do jej nadmiernej syntezy. Chromosom 11q15 Mutacja genu cykliny D1 występuje w około 18% przypadków gruczolaków przytarczyc. Jej nadmiar pobudza przejście komórki z fazy stacjonarnej (G1) do fazy syntezy DNA (S) Cdk4 i Cdk6 hipotetyczna mutacja aktywująca genów kodujących te enzymy (kinazy) dotychczas nie została opisana. Brak doniesień klinicznych
 mutacja genu (pełniącego funkcję onkogenu) kodującego cyklinę D1. Prowadzi do jej nadmiernej syntezy. Chromosom 11q15 Mutacja genu cykliny D1 występuje w około 18% przypadków gruczolaków przytarczyc. Jej nadmiar pobudza przejście komórki z fazy stacjonarnej (G1) do fazy syntezy DNA (S) Cdk4 i Cdk6 hipotetyczna mutacja aktywująca genów kodujących te enzymy (kinazy) dotychczas nie została opisana. Brak doniesień klinicznych.")
6
MECHANIZM ZWIĄZANY Z AKTYWACJĄ SZLAKU CYKLINY D1

7
JAKA JEST ETIOLOGIA NAJCZĘSTSZEJ POSTACI PIERWOTNEJ NADCZYNNOŚCI PRZYTARCZYC CZYLI POJEDYNCZEGO GRUCZOLAKA? LOH – (szacowany udział w rozwoju gruczolaka przytarczyc: 25-40%) utrata heterozygotyczności polega na skróceniu jednego z chromosomów i utracie jednego z alleli. Chromosom 11q13 i inne. Uważa się, że LOH wiąże się z utratą genów hamujących rozrost guzów. W przypadku białek z grupy hamujących rozwój guza do prawidłowego ich funkcjonowania potrzebne są oba allele. Mutacja genu meniny – (szacowany udział w rozwoju gruczolaka przytarczyc: 20-35%) Chromosom 11q13 i inne. W przypadku genu meniny wykazującej efekt hamowaniach rozwoju guza do prawidłowego funkcjonowania potrzebne są oba allele.
 utrata heterozygotyczności polega na skróceniu jednego z chromosomów i utracie jednego z alleli. Chromosom 11q13 i inne. Uważa się, że LOH wiąże się z utratą genów hamujących rozrost guzów. W przypadku białek z grupy hamujących rozwój guza do prawidłowego ich funkcjonowania potrzebne są oba allele. Mutacja genu meniny – (szacowany udział w rozwoju gruczolaka przytarczyc: 20-35%) Chromosom 11q13 i inne. W przypadku genu meniny wykazującej efekt hamowaniach rozwoju guza do prawidłowego funkcjonowania potrzebne są oba allele..")
8
UPROSZCZONY SCHEMAT DZIAŁANIA MENINY

9
CZY ROZPOZNANIE GRUCZOLAKA PRZYTARCZYC UZASADNIA PRZEPROWADZENIE BADAŃ GENETYCZNYCH? ARGUMENTY „PRZECIW”ARGUMENTY „ZA” * Gruczolaki przytarczyc rzadko ulegają transformacji złośliwej * Bardzo rzadko gruczolaki ulegają transformacji złośliwej i najprawdopodobniej odpowiedzialne są czynniki epigenetyczne jak np. nieprawidłowa metylacja, dołączenie się inne mutacji somatycznej * Gruczolak przytarczyc jest rozrostem monoklonalnym i jego resekcja przeprowadzona zgodnie ze standardem spowoduje wyleczenie * Nie istnieje metoda, która przed paratyreoidektomią mogłaby potwierdzić jednoznacznie, że zmiana wydzielająca PTH jest gruczolakiem * Wysoki koszt badania * Mutacje mają charakter somatyczny, więc badanie genetyczne nic nie wniesie. * Nadczynność przytarczyc w zespołach genetycznych może klinicznie być nie do odróżnienia * Istnieje możliwość oceny ekspresji parafibrominy w materiale histopatologicznym * W badaniu genetycznym możliwe jest wykrycie wielu chorób prowadzących do nadczynności przytarczyc.

10
HIPERPLAZJA KOMÓREK PRZYTARCZYC Hiperplazja jest poliklonalnym namnażaniem się komórek przytarczyc w odpowiedzi na czynniki środowiskowe (wtórna i trzeciorzędowa nadczynność przytarczyc) lub spowodowanym wadą genetyczną. Nie są znane nieinwazyjne metody różnicujące w sposób jednoznaczny rozrost poliklonalny i monoklonalny.

11
MEN 1. MUTACJA GENU KODUJĄCEGO BIAŁKO MENINĘ (11Q13.1). CZĘSTOŚĆ 1:30000. ROZPIĘTOŚĆ CZASU WYSTĄPIENIA SKŁADOWYCH ZESPOŁU: 5 – 90 ROKU ŻYCIA CechaKomentarz Pierwotna nadczynność przytarczyc Łagodna hiperkalcemia i umiarkowane podwyższenie poziomu PTH Może poprzedzać wystąpienie pozostałych składowych zespołu nawet na 30 lat. Jest najczęstszą endokrynopatią w MEN 1 i występuje u ponad 90 procent chorych. Cechą charakterystyczną jest dysproporcja między umiarkowaną hiperkalcemią, a dużym nasileniem zmian kostnych. Guzy neuroendokrynne trzustki i dwunastnicy występują u 75% chorych z MEN 1: Gastrinoma (>10%) Insulinoma (10%) Niewydzielające (<10%) Inne (glukagonoma, somatostatuinoma, vipoma) – rzadkie (*) Charakterystyczna hiperplazja. Gastrinoma w MEN 1: wieloogniskowość, mały rozmiar. Czynnik ryzyka zgonu. Nieskuteczne leczenie operacyjne (rola PPI). Insulinoma czynnik ryzyka przedwczesnego zgonu - wymaga leczenia radykalnego. Guzy niewydzielające oraz guzy rzadkie są czynnikami ryzyka przedwczesnego zgonu z uwagi na wysokie ryzyko ich zezłośliwienia. Guzy przysadki występują u około 40% chorych z MEN 1. od 5 roku życia do 9 dekady, 85% to makrogruczolaki). Często naciekają okoliczne struktury. Częstość występowania to kolejno: prolaktinoma, guzy niewydzielające, somatotropinoma i kortykotropinoma. Obecne też postacie mieszane wydzielające różne hormony. Prolaktinoma w MEN 1 często (44%) oporne na leczenie agonistami DA
 Insulinoma (10%) Niewydzielające (<10%) Inne (glukagonoma, somatostatuinoma, vipoma) – rzadkie (*) Charakterystyczna hiperplazja. Gastrinoma w MEN 1: wieloogniskowość, mały rozmiar. Czynnik ryzyka zgonu. Nieskuteczne leczenie operacyjne (rola PPI). Insulinoma czynnik ryzyka przedwczesnego zgonu - wymaga leczenia radykalnego. Guzy niewydzielające oraz guzy rzadkie są czynnikami ryzyka przedwczesnego zgonu z uwagi na wysokie ryzyko ich zezłośliwienia. Guzy przysadki występują u około 40% chorych z MEN 1. od 5 roku życia do 9 dekady, 85% to makrogruczolaki). Często naciekają okoliczne struktury. Częstość występowania to kolejno: prolaktinoma, guzy niewydzielające, somatotropinoma i kortykotropinoma. Obecne też postacie mieszane wydzielające różne hormony. Prolaktinoma w MEN 1 często (44%) oporne na leczenie agonistami DA.")
12
FIBROMA W MEN 1

13
MEN 4. MUTACJA GENU CDKN1B KODUJĄCEGO BIAŁKO P27 BĘDĄCYM INHIBITOREM KINAZY CDK2. (12Q13.1). CZĘSTOŚĆ: 3% PRZYPADKÓW OPISANYCH JAKO MEN 1. CechaKomentarz Pierwotna nadczynność przytarczyc Występuje we wszystkich dotychczas opisanych przypadkach zespołów MEN 4 ale jej obraz kliniczny jest bardzo zróżnicowany. Guzy neuroendokrynne trzustki i dwunastnicy Mała ilość opisanych przypadków. Opisano występowanie guzów neuroendokrynnych podobne jak w MEN 1 Guzy przysadkiMała liczba opisów przypadków. Dotychczas zespołowi MEN 4 towarzyszyły: kortykotropinoma, somatotropinoma i guzy nieaktywne hormonalnie.
. CZĘSTOŚĆ: 3% PRZYPADKÓW OPISANYCH JAKO MEN 1. CechaKomentarz Pierwotna nadczynność przytarczyc Występuje we wszystkich dotychczas opisanych przypadkach zespołów MEN 4 ale jej obraz kliniczny jest bardzo zróżnicowany. Guzy neuroendokrynne trzustki i dwunastnicy Mała ilość opisanych przypadków. Opisano występowanie guzów neuroendokrynnych podobne jak w MEN 1 Guzy przysadkiMała liczba opisów przypadków. Dotychczas zespołowi MEN 4 towarzyszyły: kortykotropinoma, somatotropinoma i guzy nieaktywne hormonalnie..")
14
MECHANIZM TŁUMACZĄCY PODOBIEŃSTWO OBRAZU KLINICZNEGO MEN1 I MEN4

15
MEN2A. MUTACJA AKTYWUJĄCA ONKOGEN RET (ARRANGED DURING TRANSFECTION). 10Q11.21 GEN KODUJE DUŻE BIAŁKO: PRZEZBŁONOWY RECEPTOR KINAZY TYROZYNOWEJ Nadczynność przytarczycPojawia się ona u 25% chorych, najczęściej w czwartej dekadzie życia, ma charakter łagodny. Wykazuje nawrotowośc jak w MEN1, dlatego przy okazji tyreoidektomii wykonuje się subtotalną paratyreoidektomię i tymektomię Guz chromochłonnyklinicznego ujawnienia się w zespole MEN2A przypada na czwartą dekadę życia. W formie utajonej może występować dużo wcześniej i przyczynić się do zgonów śródoperacyjnych. Rak rdzeniasty tarczycyNajczęstsza endokrynopatia. Agresywność zależna od lokalizacji mutacji

16
MUTACJA GENU CDC73 [HRPT2] (1Q31.2) GEN KODUJE BIAŁKO PARAFIBROMINĘ PEŁNIĄCE FUNKCJĘ CZYNNIKA HAMUJĄCEGO ROZWÓJ GUZA PostaćKomentarz Zespół HPT-JT (ang. Hyperparathyroidism- jaw tumor) Rozpoznanie: Pierwotna nadczynność przytarczyc i jednej lub więcej z poniższych chorób: Kostniejący włókniak szczęki lub żuchwy Zespół HPT-JT wśród najbliższych krewnych. lub występowanie kostniejących włókniaków żuchwy lub szczęki i zespół HPT-JT wśród najbliższych krewnych. CDC73 zależna rodzinna, izolowana nadczynność przytarczyc (FIHP - ang. CDC73 related familial isolated hyperparathyroidism) Rozpoznanie: nadczynność przytarczyc, obecność mutacji germinalnej (wrodzonej) genu CDC73, występowanie nadczynności przytarczyc bez zmian kostnych u członka najbliższej rodziny Rak przytarczyc w przebiegu mutacji CDC73. (CDC73-related parathyroid carcinoma). Rozpoznanie: rak przytarczyc i wrodzona, patogenna mutacja CDC73
![MUTACJA GENU CDC73 [HRPT2] (1Q31.2) GEN KODUJE BIAŁKO PARAFIBROMINĘ PEŁNIĄCE FUNKCJĘ CZYNNIKA HAMUJĄCEGO ROZWÓJ GUZA PostaćKomentarz Zespół HPT-JT (ang.](http://images.slideplayer.pl/41/11508735/slides/slide_16.jpg "Hyperparathyroidism- jaw tumor) Rozpoznanie: Pierwotna nadczynność przytarczyc i jednej lub więcej z poniższych chorób: Kostniejący włókniak szczęki lub żuchwy Zespół HPT-JT wśród najbliższych krewnych. lub występowanie kostniejących włókniaków żuchwy lub szczęki i zespół HPT-JT wśród najbliższych krewnych. CDC73 zależna rodzinna, izolowana nadczynność przytarczyc (FIHP - ang. CDC73 related familial isolated hyperparathyroidism) Rozpoznanie: nadczynność przytarczyc, obecność mutacji germinalnej (wrodzonej) genu CDC73, występowanie nadczynności przytarczyc bez zmian kostnych u członka najbliższej rodziny Rak przytarczyc w przebiegu mutacji CDC73. (CDC73-related parathyroid carcinoma). Rozpoznanie: rak przytarczyc i wrodzona, patogenna mutacja CDC73.")
17
GERMINALNA MUTACJA CDC73

18
MUTACJA CDC73 GERMINALNA (PO LEWEJ) I SOMATYCZNA (PO PRAWEJ)
 I SOMATYCZNA (PO PRAWEJ)")
19
KIEDY PODEJRZEWAĆ GENETYCZNE PODŁOŻE NADCZYNNOŚCI PRZYTARCZYC CZYLI KIEDY PONOWNIE ROZWAŻYĆ BADANIE GENETYCZNE? Cecha klinicznaKomentarz Nadczynność przytarczyc w młodym (<40roku życia) wieku Cecha spotykana najczęściej w zespołach MEN 1 i 4 oraz raku przytarczyc Wartości PTH 3 x powyżej górnej granicy normy Cecha spotykana we wtórnej i trzeciorzędowej nadczynności przytarczyc oraz raku przytarczyc Kalcemia > 3mmol/lPodejrzenie raka przytarczyc Zaawansowane zmiany kostne – guzy brunatne, guzy żuchwy Cecha charakterystyczna dla germinalnych mutacji CDC73 Nieadekwatnie zaawansowane zmian kostnych w stosunku do łagodnej nadczynności przytarczyc Cecha spotykana w MEN 1 Cechy naciekania okolicznych tkanek w USG/MIBI Groźny objaw spotykany w raku przytarczyc Nawrotowość nadczynności przytarczycCecha typowa dla MEN 1, 2A, 4 oraz spotkana w mutacjach CDC73
 wieku Cecha spotykana najczęściej w zespołach MEN 1 i 4 oraz raku przytarczyc Wartości PTH 3 x powyżej górnej granicy normy Cecha spotykana we wtórnej i trzeciorzędowej nadczynności przytarczyc oraz raku przytarczyc Kalcemia > 3mmol/lPodejrzenie raka przytarczyc Zaawansowane zmiany kostne – guzy brunatne, guzy żuchwy Cecha charakterystyczna dla germinalnych mutacji CDC73 Nieadekwatnie zaawansowane zmian kostnych w stosunku do łagodnej nadczynności przytarczyc Cecha spotykana w MEN 1 Cechy naciekania okolicznych tkanek w USG/MIBI Groźny objaw spotykany w raku przytarczyc Nawrotowość nadczynności przytarczycCecha typowa dla MEN 1, 2A, 4 oraz spotkana w mutacjach CDC73.")
20
KIEDY PODEJRZEWAĆ GENETYCZNE PODŁOŻE NADCZYNNOŚCI PRZYTARCZYC CZYLI KIEDY PONOWNIE ROZWAŻYĆ BADANIE GENETYCZNE? Cecha klinicznakomentarz Współwystępowanie innych endokrynopatii, w tym gruczolaków przysadki wydzielających kilka hormonów tropowych Cecha zespołu MEN 1 Torbiele nerekCecha w mutacji CDC73 Gruczolaki nadnerczaCecha towarzysząca MEN1 Przytarczyca wyczuwalna w badaniu palpacyjnym Rzadki objaw raka przytarczyc Niewystępowanie hiperkalciuriiMożliwość występowania łagodnej hiperkalcemii hipokalciurycznej (mutacji inaktywującej receptor wapniowy) Powiększenie więcej niż jednej przytarczycy w badaniach obrazowych Pośredni dowód na germinalny charakter choroby Zwapnienia przytarczycy w USGMoże odpowiadać ogniskom martwicy (w raku przytarczyc)
 Powiększenie więcej niż jednej przytarczycy w badaniach obrazowych Pośredni dowód na germinalny charakter choroby Zwapnienia przytarczycy w USGMoże odpowiadać ogniskom martwicy (w raku przytarczyc).")
21
CZY WYNIK BADANIA GENETYCZNEGO BĘDZIE MIAŁ WPŁYW NA POSTĘPOWANIE TERAPEUTYCZNO-DIAGNOSTYCZNE? RozpoznanieDecyzja terapeutyczno-diagnostyczna Rodzinna hiperkalcemia hipokalciuryczna FHH Diagnostyka biochemiczna zawodzi zwłaszcza w niewydolności nerek, w przypadkowym stosowaniu diuretyku tiazydowego, w kamicy nerkowej. Rozpoznanie choroby zmienia leczenie, które się indywidualizuje. MEN 1Podstawa do nadzoru endokrynologicznego i radykalnego leczenia. Ważna rola poradnictwa genetycznego MEN 2AJak wyżej MEN 4Jak wyżej CDC 73Podstawa do nadzoru i indywidualizacji leczenia, chirurgia minimalnie inwazyjna.

22
HIPOKALCEMIA U OSOBY BEZ WYWIADU LECZENIA CHIRURGICZNEGO ANI RADIOTERAPII W OBRĘBIE SZYI. To musi być choroba o podłożu genetycznym Przyczyny: Nieprawidłowy rozwój przytarczyc (mutacja GCM2, del 10p13- 14, del 22q11, mutacja TBCE, mutacja FAM111A) Odpowiedź immunologiczną skierowana przeciw przytarczycom (AIRE) Uszkodzenie genu odpowiedzialnego za syntezę PTH Odporność na działanie PTH (mutacje i zaburzenia metylacji GNAS 1) Zaburzenia regulacji wydzielania PTH (mutacja CASR)
 Odpowiedź immunologiczną skierowana przeciw przytarczycom (AIRE) Uszkodzenie genu odpowiedzialnego za syntezę PTH Odporność na działanie PTH (mutacje i zaburzenia metylacji GNAS 1) Zaburzenia regulacji wydzielania PTH (mutacja CASR).")
23
HIPOKALCEMIA U OSOBY BEZ WYWIADU LECZENIA CHIRURGICZNEGO ANI RADIOTERAPII W OBRĘBIE SZYI. Większość tych chorób zostaje wykryta w dzieciństwie, jednak różna penetracja genów oraz charakter mutacji powodują, że choroba może ujawnić się dopiero w wieku dorosłym. Przykładem są: Mutacja genu GCM2 Mikrodelecja 22q11 Mutacja aktywująca CASR Rzekoma niedoczynność przytarczyc IB APS 1, APS 3 i APS 4

24
PODZIAŁ AUTOIMMUNOLOGICZNYCH ZESPOŁÓW WIELOGRUCZOŁOWYCH (APS) PRZEBIEGAJĄCYCH Z NIEDOCZYNNOŚCIĄ PRZYTARCZYC Typ zespołu APS → ↓ Objaw APS 1APS 3APS 4 Niedoczynność przytarczyc TAK Niedoczynność kory nadnerczy TAK NIE Autoimmunologiczna niedoczynność tarczycy NIETAKNIE Inne choroby z autoagresji TAK
 PRZEBIEGAJĄCYCH Z NIEDOCZYNNOŚCIĄ PRZYTARCZYC Typ zespołu APS → ↓ Objaw APS 1APS 3APS 4 Niedoczynność przytarczyc TAK Niedoczynność kory nadnerczy TAK NIE Autoimmunologiczna niedoczynność tarczycy NIETAKNIE Inne choroby z autoagresji TAK")
25
KLINICZNE KRYTERIA APS 1: 1) NIEDOCZYNNOŚĆ PRZYTARCZYC, 2) KANDYDOZA, 3) CHOROBA ADDISONA ORAZ DWA Z NASTĘPUJĄCYCH OBJAWÓW TOWARZYSZĄCYCH: CUKRZYCA TYPU 1, PIERWOTNY HIPOGONADYZM, AUTOIMMUNOLOGICZNE ZAPALENIE WĄTROBY, ŁYSIENIE PLACKOWATE I BIELACTWO Mutacja → ↓ Objaw c.796C>T Centr. i Płn. Europa, tzw. „Fińska” c.967-979del13 „Norwesko-angielsko- amerykańska” c.254A>G „Irańska” c.415>T „Sardyńska” Niedoczynność przytarczyc 79%82%96%93% Niedoczynność kory nadnerczy 72%68%22%73% Kandydoza błon śluzowych 100%79%17%83% Autoimmunologiczne zapalenie wątroby 12%3%?20% Łysienie plackowate 29%41%13%37% Bielactwo 13%20%?12% Cukrzyca typu 1 12%9%4%2.5%

26
PRZYCZYNY ZGONU W APS 1 * Powikłania grzybicy przewodu pokarmowego jak rak przełyku. * Niewydolność wątroby w przypadku jej autoimmunologicznego zapalenia. * Przełom nadnerczowy. * Ciężka hipokalcemia. * Powikłania infekcyjne (w przypadku hipotrofii śledziony w APS 1 zaleca się szczepienia przeciw pneumokokom).
..")
27
MIKRODELECJA 22Q11 Występuje z częstością 1 na 2000 żywo urodzonych dzieci! Hipokalcemia może być niestała ale w razie wystąpienia, objawy mogą być nasilone. Stanowi czynnik etiologiczny schizofrenii i zespołów depresyjnych. Epizody hipokalcemii mogą pogarszać przebieg chorób psychiatrycznych.

28
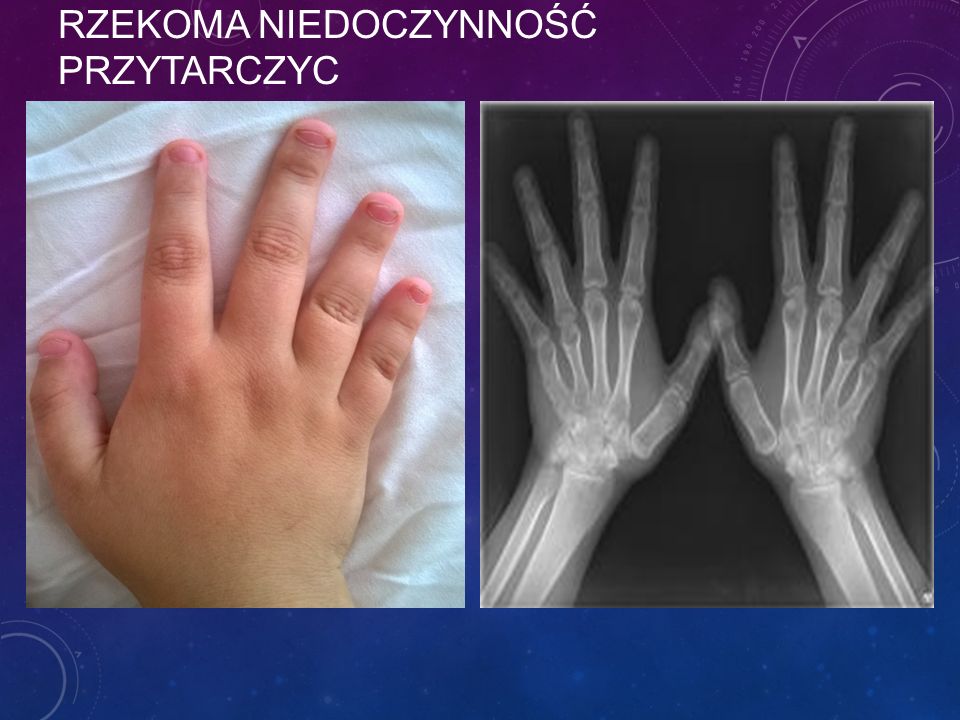
RZEKOMA NIEDOCZYNNOŚĆ PRZYTARCZYC

30
RZEKOMA NIEDOCZYNNOŚĆ PRZYTARCZYC TYPU 1B

31
CZY BADANIE GENETYCZNE U CHORYCH Z NIEDOCZYNNOŚCIĄ PRZYTARCZYC ZMIENI POSTĘPOWANIE TERAPEUTYCZNE? PatologiaZastosowanie kliniczne APS 1Badanie genetyczne jest konieczne do rozpoznania, indywidualizacji leczenia i rokowania (nowe leki) Delecja 22q11Konieczna diagnostyka składowych (poronnych postaci) zespołu Di George, duże znaczenie w poradnictwie genetycznym (ciąże nie są zalecane) Mutacja aktywująca receptor wapniowy Indywidualizacja leczenia, zwłaszcza pod kątem późnych powikłań. PozostałePoradnictwo genetyczne zwłaszcza w postaciach objawowych.
 Delecja 22q11Konieczna diagnostyka składowych (poronnych postaci) zespołu Di George, duże znaczenie w poradnictwie genetycznym (ciąże nie są zalecane) Mutacja aktywująca receptor wapniowy Indywidualizacja leczenia, zwłaszcza pod kątem późnych powikłań. PozostałePoradnictwo genetyczne zwłaszcza w postaciach objawowych..")
32
PROPONOWANE BADANIA PIERWSZEGO RZUTU GENETYCZNE U OSÓB Z PODEJRZENIEM GENETYCZNEGO PODŁOŻA ZABURZEŃ FUNKCJI PRZYTARCZYC (HIPREKALCEMIA) MEN 1 (11q31.1)Zespół MEN 1 CDKN1B (12q13.1)Zespół MEN 4 RET (10q11.21)Zespół MEN 2A CDC73 (1q31.2) /HRPT 2HPT-JT/FIHP/rak przytarczyc CASR (3q13.3-3q21.1)FHH typ 1 (mutacja inaktywująca) GNA11 (19p31.3)FHH typ 2 (mutacja inaktywująca) AP2S1 (19q13.32)FHH typ 3 (mutacja inaktywująca)
 MEN 1 (11q31.1)Zespół MEN 1 CDKN1B (12q13.1)Zespół MEN 4 RET (10q11.21)Zespół MEN 2A CDC73 (1q31.2) /HRPT 2HPT-JT/FIHP/rak przytarczyc CASR (3q13.3-3q21.1)FHH typ 1 (mutacja inaktywująca) GNA11 (19p31.3)FHH typ 2 (mutacja inaktywująca) AP2S1 (19q13.32)FHH typ 3 (mutacja inaktywująca)")
33
PROPONOWANE BADANIA GENETYCZNE PIERWSZEGO RZUTU U OSÓB Z PODEJRZENIEM GENETYCZNEGO PODŁOŻA ZABURZEŃ FUNKCJI PRZYTARCZYC (HIPOKALCEMIA) Proponowana diagnostyka genetycznaJednostka chorobowa del 22q11DiGeorgi, postacie łagodne AIRE (21q22.3)APS 1 GNAS (20q13.2-13.3)Rzekoma niedoczynność przytarczyc (zwłaszcza 1B) CASR (3q13.3-3q21.1)HYPOC 1 (mutacja aktywująca) GNA11 (19p31.3)HYPOC 2 (mutacja aktywująca)
 Proponowana diagnostyka genetycznaJednostka chorobowa del 22q11DiGeorgi, postacie łagodne AIRE (21q22.3)APS 1 GNAS (20q )Rzekoma niedoczynność przytarczyc (zwłaszcza 1B) CASR (3q13.3-3q21.1)HYPOC 1 (mutacja aktywująca) GNA11 (19p31.3)HYPOC 2 (mutacja aktywująca)")
34
PODSUMOWANIE Czy gruczolak przytarczyc może ulec przemianie nowotworowej? Tak, ale nie wydaje się aby decydował o tym tylko czas trwania choroby. W tym „czasie” muszą zaistnieć sprzyjające warunki. Czy dochodzące do 5% występowanie raka przytarczyc może być niedoszacowane? Prawdopodobnie tak. Czy można „obserwować” nadczynność przytarczyc? Tak, ale należy monitorować zwłaszcza te cechy kliniczne, które mogą przemawiać za procesem złośliwym. Czy niedoczynność przytarczyc jest rzadką chorobą? Nie! Czy wynik badania genetycznego w niedoczynności przytarczyc wpływa na dalsze postępowanie z Chorym? Tak! Zwłaszcza w delecji 22q11 i APS.




 ogół rzeczywistych jednostek, o których chcemy uzyskać informacje.>")




 do zakresu komórek w innym skoroszycie Możliwości efektywnego stosowania odwołań zewnętrznych Odwołania zewnętrzne.>")





. Powszechnie znane typy cukrzycy Typ I Cukrzyca typu 1 występuje wtedy, gdy własny układ odpornościowy organizmu niszczy.>")





