Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Mutacje pojedynczego genu – choroby jednogenowe
Lek. med. Anna Kruczek Kurs „Podstawy genetyki klinicznej” Kraków,

2
ZESPÓŁ MARFANA Wieloukładowa choroba tkanki łącznej o dużej zmienności fenotypowej – zmiany narządowe dotyczą: Układu kostno-stawowego Układu krążenia (serce i naczynia krwionośne) Narządu wzroku Częstość występowania 1-2:10 tys. żywych urodzeń
 Narządu wzroku. Częstość występowania 1-2:10 tys. żywych urodzeń.")
3
ZESPÓŁ MARFANA Dziedziczenie: AD Gen: FBN1
Produkt genu: białko fibrylina 1 – główny składnik miofibryli tkanki łącznej Locus genowe: 15q21.1 Duży stopień penetracji i zmienna ekspresja genu 70-85% przypadków rodzinnych

4
OBJAWY – UKŁAD KOSTNO-STAWOWY
Wysoki wzrost – zwykle powyżej 97 percentyla (mężczyźni 191,3 ± 9cm, kobiety 175,4 ± 8,2cm) Zaburzenia proporcji ciała Dolichostenomelia Wymiar górny do dolnego poniżej 0,85 Sięg do wzrostu >1,05 Nieprawidłowa budowa klatki piersiowej Wady kręgosłupa Arachnodaktylia Objaw nadgarstkowy (Walkera-Murdocha) Objaw kciuka (Steinberga)
 Zaburzenia proporcji ciała. Dolichostenomelia. Wymiar górny do dolnego poniżej 0,85. Sięg do wzrostu >1,05. Nieprawidłowa budowa klatki piersiowej. Wady kręgosłupa. Arachnodaktylia. Objaw nadgarstkowy (Walkera-Murdocha) Objaw kciuka (Steinberga)")
5
OBJAWY – UKŁAD KOSTNO-STAWOWY
Wady stóp (długie i wąskie, stopa wydrążona, stopy płasko-koślawe, palce młotkowate, rotacja przyśrodkowa kostki przyśrodkowej) Nadmierna ruchomość stawów lub przykurcze Genu recurvatum
 Nadmierna ruchomość stawów lub przykurcze. Genu recurvatum.")
6
OBJAWY – UKŁAD SERCOWO-NACZYNIOWY
Poszerzenie opuszki aorty lub pnia płucnego Wypadanie płatków zastawek (mitralna, trójdzielna) Niedomykalność zastawki (aortalnej, mitralnej) Tętniak rozwarstwiający aorty Zastoinowa niewydolność krążenia
 Niedomykalność zastawki (aortalnej, mitralnej) Tętniak rozwarstwiający aorty. Zastoinowa niewydolność krążenia.")
7
OBJAWY – NARZĄD WZROKU Wady soczewki (luxatio, subluxatio) Enophtalmus
Krótkowzroczność Wady budowy gałki ocznej Zwiększony wymiar osi długiej Płaska rogówka Hipoplazja tęczówki Jaskra, zaćma Odwarstwienie siatkówki Niebieskie twardówki

8
OBJAWY - DYSMORFIA Dolichocefalia Długa, wąska twarz
Antymongoidalne ustawienie szpar powiekowych Hipoplazja policzków Mała, cofnięta żuchwa Gotyckie podniebienie Stłoczenie zębów

9
OBJAWY - INNE Układ oddechowy – rozedma, płuc, samoistne pęcherze rozedmowe, odma Przepukliny Obniżona masa mięśniowa Skąpa podskórna tkanka tłuszczowa Rozstępy Zwyrodnienie stawów Poszerzenie worka opony twardej w odcinku lędźwiowo-krzyżowym (dural ectasia)
")
10
ROZPOZNANIE – SKALA PUNKTOWA
Dodatni objaw nadgarstkowy i/lub kciuka /1 Klatka piersiowa kurza lub szewska/asymetryczna 2/1 Stopa płasko-koślawa lub płaskostopie /1 Odma opłucnowa Dural ectasia Protruzja panewki

11
ROZPOZNANIE – SKALA PUNKTOWA
Zmniejszony stosunek segm. górnego do dolnego i zwiększony sięg do wysokości 1 Skolioza lub kifoza w odcinku piersiowo-lędźwiowym 1 Ograniczenie wyprostu w stawach łokciowych 1 Cechy dysmorfii (3 z 5: długa, wąska twarz; enophtalmos; antymongoidalne ustawienie szpar powiekowych; hipoplazja policzków; mała, cofnięta żuchwa) 1 Rozstępy skórne Krótkowzroczność Wypadanie płatka zastawki mitralnej 1
 1. Rozstępy skórne 1. Krótkowzroczność 1. Wypadanie płatka zastawki mitralnej 1.")
12
KRYTERIA ROZPOZNANIA Nieobciążony wywiad rodzinny
1) Poszerzenie aorty u osób poniżej 20 roku życia – Z-score ≥3.0 u osób powyżej 20 roku życia – Z-score ≥2.0 I jedno z poniższych Ektopia soczewki Patogenna mutacja w genie FBN1 Skala punktowa – 7 pkt. lub więcej 2) Ektopia soczewki i mutacja w genie FBN1, dla której opisano związek z poszerzeniem aorty
 Poszerzenie aorty u osób poniżej 20 roku życia – Z-score ≥3.0 u osób powyżej 20 roku życia – Z-score ≥2.0. I jedno z poniższych. Ektopia soczewki. Patogenna mutacja w genie FBN1. Skala punktowa – 7 pkt. lub więcej. 2) Ektopia soczewki i mutacja w genie FBN1, dla której opisano związek z poszerzeniem aorty.")
13
KRYTERIA ROZPOZNANIA Obciążony wywiad rodzinny i jedno z poniższych
Ektopia soczewki Skala punktowa – ≥7 pkt. Poszerzenie aorty

14
ROZPOZNANIE U DZIECI I MŁODZIEŻY
Niespecyficzna choroba tkanki łącznej Skala punktowa <7 Graniczna szerokość aorty (Z-score <3.0) Brak mutacji Potencjalny zespół Marfana Znana mutacja
 Brak mutacji. Potencjalny zespół Marfana. Znana mutacja.")
15
BADANIA MOLEKULARNE Opisano ponad 600 mutacji genu FBN1
Identyfikacja mutacji jest jednym z kryteriów diagnostycznych Brak identyfikacji mutacji nie wyklucza rozpoznania klinicznego

16
BADANIA ECHO serca – poszerzenie aorty jako główne kryterium diagnostyczne Badanie okulistyczne – ektopia soczewki Pomiary antropometryczne Konsultacja ortopedyczna (RTG)
")
17
CHOROBY ALLELICZNE Zespół wypadania płatków zastawki mitralnej (z nieprawidłowościami układu kostnego lub bez) Fenotyp MASS – myopia, mitral valve prolapse, aortic enlargment (niepostępujące), skin and skeletal features (niespecyficzne) Rodzinne zwichnięcie soczewki Zespół Weilla-Marchesaniego – zwichnięcie soczewki, microspherophakia, niedobór wzrostu, brachydaktylia, brak objawów z układu krążenia Zespół Loeysa-Dietza – większość objawów typowych dla zespołu Marfana bez zwichnięcia soczewek
, skin and skeletal features (niespecyficzne) Rodzinne zwichnięcie soczewki. Zespół Weilla-Marchesaniego – zwichnięcie soczewki, microspherophakia, niedobór wzrostu, brachydaktylia, brak objawów z układu krążenia. Zespół Loeysa-Dietza – większość objawów typowych dla zespołu Marfana bez zwichnięcia soczewek.")
18
DIAGNOSTYKA RÓŻNICOWA
Homocystynuria Cechy wspólne: dolichostenomelia, dolichocefalia, arachnodaktylia, deformacje klatki piersiowej, skolioza, wady oczne (podwichnięcie soczewek, krótkowzroczność, zaćma, jaskra) Różnice: przykurcze stawowe, brak objawów ze strony serca, zakrzepica, deficyty intelektualne, padaczka, dziedziczenie AR
 Różnice: przykurcze stawowe, brak objawów ze strony serca, zakrzepica, deficyty intelektualne, padaczka, dziedziczenie AR.")
19
DIAGNOSTYKA RÓŻNICOWA
Zespół Ehlersa-Danlosa – wiotkość stawów, niebieskie twardówki, kifoskolioza i podwichnięcie soczewek (typ VI) Marfanoidalny zespół nadmiernej ruchomości stawów (Marfanoid hypermobility syndrome) MFS – zmiany w układzie kostno-stawowym i krążenia, bez wad ocznych EDS – nadmierna elastyczność skóry, tendencja do wybroczyn, trudne gojenie ran, skóra pergaminowa
 Marfanoidalny zespół nadmiernej ruchomości stawów (Marfanoid hypermobility syndrome) MFS – zmiany w układzie kostno-stawowym i krążenia, bez wad ocznych EDS – nadmierna elastyczność skóry, tendencja do wybroczyn, trudne gojenie ran, skóra pergaminowa.")
20
DIAGNOSTYKA RÓŻNICOWA
Zespół Shprintzena-Goldberga – dolichostenomelia, arachnodaktylia, deformacje klatki piersiowej, skolioza oraz hiperteloryzm, kraniosynostoza, zaburzenia poznawcze Zespół Lejuna-Frynsa – niepełnosprawność intelektualna z cechami marfanoidalnymi sprzężona z chromosomem X Zespół łamliwego chromosomu X Zespół Klinefeltera

21
OPIEKA NAD PACJENTEM Porada genetyczna typowa dla choroby AD
Opieka wielospecjalistyczna: kardiolog, okulista, ortopeda Rehabilitacja Unikanie nadmiernego wysiłku z przyczyn kardiologicznych Leczenie prewencyjne przed zabiegami stomatologicznymi Właściwy wybór zawodu

22
NEUROFIBROMATOZA TYPU I
Fakomatoza (gr. phakos – naznaczony przy urodzeniu) Choroba wieloukładowa – objawy głównie z zakresu neuroektodermy Skóra Układ nerwowy Narząd wzroku Częstość występowania 1:3000 żywych urodzeń (jedna z najczęściej występujących chorób o AD modelu dziedziczenia, bardzo często pozostaje nierozpoznana)
 Choroba wieloukładowa – objawy głównie z zakresu neuroektodermy. Skóra. Układ nerwowy. Narząd wzroku. Częstość występowania 1:3000 żywych urodzeń (jedna z najczęściej występujących chorób o AD modelu dziedziczenia, bardzo często pozostaje nierozpoznana)")
23
NF-1 Dziedziczenie: AD Gen: NF1
Produkt genu: białko neurofibromina – regulator aktywności onkogenu p21ras i niektórych czynników wzrostowych Locus genowe: 17q11.2 Penetracja sięga 100%, bardzo zmienna ekspresja genu 50% przypadków rodzinnych

24
KRYTERIA DIAGNOSTYCZNE
Opracowane w 1997 jako tzw. NF1 NIH Consensus Conference Criteria Plamy „cafe-au-lait” – 6 lub więcej plam większych niż 5 mm przed okresem dojrzewania i 15 mm po okresie dojrzewania Pojedyncze plamy występują u 10-15% zdrowej populacji. Piegi lub przebarwienia w niedostępnych dla światła okolicach ciała (pachy, wzgórek łonowy)
")
25
OBJAWY WIODĄCE Nerwiakowłókniaki – 2 lub więcej jakiegokolwiek typu lub 1 nerwiak splotowaty Włókniaki skórne zlokalizowane są w obrębie skóry i naskórka, mają „gumowatą” konsystencję i można je w niewielkim stopniu przemieszczać. Są niebolesne, czasem swędzące. Z wiekiem ewoluują do form brodawkowatych. Duże guzki podskórne mogą osiągać rozmiary kilku cm, z reguły rosną szybko w pierwszych latach życia, potem ulegają stabilizacji. Skóra nad nimi może być znacznie zmieniona. Może im towarzyszyć ból, świąd i inne objawy ucisku na nerwy lub kości. Czasem ulegają transformacji nowotworowej. Nerwiaki splotowate mogą obejmować pochewki wieku nerwów i ich odgałęzień.

26
OBJAWY WIODĄCE Glejak narządu wzroku Glejak narządu wzroku i mózgu oraz tzw. ogniska zwiększonej intensywności sygnału T2 w badaniu MRI występują u około 15% pacjentów. Mają zwykle strukturę astrocytoma pilocyticum. Mogą być zlokalizowane na całym przebiegu drogi wzrokowej, co wywołuje objawy: proptosis, obniżenie ostrości wzroku, ubytki pola widzenia. Czasem objawem jest padaczka. Lokalizacja podwzgórzowa odpowiedzialna jest za endokrynopatie: niedobór hormonu wzrostu, przedwczesne pokwitanie. Niekiedy dochodzi do samoistnej regresji zmian lub ich stabilizacji – jest to trudne do przewidzenia.

27
OBJAWY WIODĄCE Guzki Lischa – 2 lub więcej Hamartomata tęczówki – mają znaczenie diagnostyczne, nie stanowią zagrożenia dla narządu wzroku. Charakterystyczne objawy kostne- skolioza i dysplazja kości klinowej lub kości długich, osteopenia i osteoporoza. Obciążony wywiad rodzinny - chory krewny pierwszego stopnia

28
KRYTERIA DIAGNOSTYCZNE
Dorośli – dwa lub więcej spośród wymienionych objawów Dzieci 50% chorych dzieci spełnia kryteria NIH w wieku roku, w wieku 8 lat prawie wszystkie U dzieci z dodatnim wywiadem rodzinnym wystarczy jedno kryterium diagnostyczne dla rozpoznania NF1 (najczęściej są to plamy „cafe-au-lait”) Dzieci z licznymi plamami bez innych objawów klinicznych i bez obciążonego wywiadu rodzinnego powinny być uważnie obserwowane i regularnie badane (również okulistycznie)
 Dzieci z licznymi plamami bez innych objawów klinicznych i bez obciążonego wywiadu rodzinnego powinny być uważnie obserwowane i regularnie badane (również okulistycznie)")
29
OBJAWY - INNE Wady naczyniowe
Zwężenie tętnic nerkowych prowadzące do nadciśnienia Koarktacja aorty Zwężenie zastawki tętnicy płucnej Angiopatie naczyń mózgowych Deficyty psychospołeczne – występują u 1/3 pacjentów, ale NI znacznego stopnia dotyczy mniej niż 1% chorych. Zaburzenia orientacji wzrokowo-przestrzennej Dysleksja Gorsza pamięć krótkoterminowa Gorsze wyniki testów słownych w badaniach II.

30
NOWOTWORY Mięsaki prążkowanokomórkowe (rhabdomyosarcoma)
Złośliwe guzy otoczki nerwów obwodowych (MPNST – Malignant Peripheral Nerve Sheath Tumors) – najczęściej powstają na obszarze włókniaków splotowatych i są wysoce agresywne. Gwiaździaki (astrocytoma) Białaczki Rzadziej: pheochromocytoma, melanoma
 – najczęściej powstają na obszarze włókniaków splotowatych i są wysoce agresywne. Gwiaździaki (astrocytoma) Białaczki. Rzadziej: pheochromocytoma, melanoma.")
31
BADANIA MOLEKULARNE W obrębie genu NF1 stwierdza się wszystkie rodzaje mutacji – w większości powodują one przedwczesną terminację syntezy białka 90% - mutacje jednopunktowe 5% - delecje/duplikacje wewnątrzgenowe <1% - duże rearanżacje

32
BADANIA Ocena dermatologiczna najlepiej z dokumentacją fotograficzną
Badanie okulistyczne w lampie szczelinowej – guzki Lischa Konsultacja kardiologiczna – pomiar ciśnienia i ewentualne badanie ECHO serca, USG tętnic nerkowych lub angiografia Badanie neurologiczne i MRI Konsultacja ortopedyczna – skolioza, dysplazja, zmniejszona gęstość kości

33
INNE FORMY NF1 NF1 segmentowa – objawy zlokalizowane asymetrycznie i tylko w niektórych okolicach ciała, często wzdłuż linii wzrostowych Blaschko, z reguły nie ma objawów ze strony OUN ani guzków Lischa. Mutacje w genie NF1 stwierdza się tylko w materiale pobranym z obszarów zmienionych chorobowo. Postać ma charakter zmiany de novo. NF1-Noonan – fenotyp ZN (hiperteloryzm, antymongoidalne ustawienie szpar powiekowych, nisko osadzone uszy, szeroka szyja, stenoza płuca). U części pacjentów stwierdza się mutacje w genie NF1 lub PTPN11.
. U części pacjentów stwierdza się mutacje w genie NF1 lub PTPN11.")
34
INNE FORMY NF1 Zespół LEOPARD L – plamy soczewicowate (lentiginosis)
E – nieprawidłowy zapis EKG spowodowany kardiomiopatią przerostową O – ocular hypertelorism P – pulmonar stenosis A – abnormal genitalia R – retardation of growth D – deafness U większości chorych przyczyną choroby jest mutacja w genie PTPN11, ale w pojedynczych przypadkach zidentyfikowano mutacje w genie NF1.

35
DIAGNOSTYKA RÓŻNICOWA
Zespół Legiusa – liczne plamy „cafe-au-lait”, piegi pod pachami, wielkogłowie i niekiedy cechy dysmorfii odpowiadające zespołowi Noonana. Rzadko występują guzki Lischa, nerwiakowłókniaki czy guzy OUN. Przyczyną jest mutacja dominująca genu SPRED1. Mnogie plamy „cafe-au-lait” (OMIM ) Zespół McCune-Albright – zmiany barwnikowe duże, o nieregularnych brzegach, dysplazja włóknista kości, zaburzenia endokrynologiczne (przedwczesne dojrzewanie, nadczynność tarczycy i kory nadnerczy)
 Zespół McCune-Albright – zmiany barwnikowe duże, o nieregularnych brzegach, dysplazja włóknista kości, zaburzenia endokrynologiczne (przedwczesne dojrzewanie, nadczynność tarczycy i kory nadnerczy)")
36
OPIEKA NAD PACJENTEM Porada genetyczna typowa dla choroby AD
Opieka wielospecjalistyczna Pomiar ciśnienia tętniczego 1 x w miesiącu Kontrola auksologiczna 1 x w roku Kontrola ortopedyczna, dermatologiczna, okulistyczna, neurologiczna 1 x w roku lub w zależności od stwierdzonych objawów MRI co 2 lata Ocena ogólnorozwojowa – psycholog, logopeda Przeżycie – średnia długość życia jest o 8 lat krótsza niż w zdrowej populacji. Przyczyną wcześniejszych zgonów są nowotwory (MPNST) i anomalie naczyniowe.
 i anomalie naczyniowe.")
37
MIKRODELECJA 17q11.1 Cechy dysmorfii (90%): makrocefalia, hiperteloryzm, ptoza, rozszczep tęczówki, mikrognatia Wysoki wzrost (46%) Duże dłonie i stopy (46%) Skolioza (43%) Nadmierna ruchomość stawów (72%) Opóźnienie rozwoju psychoruchowego, problemy szkolne (93%), zaburzenia koncentracji (73%), opóźnienie rozwoju mowy (48%), NI (38%) Wcześniejsze wystąpienie objawów typowych dla NF-1 (przed 5 rokiem życia) Nerwiakowłókniaki (76%) Nerwiaki splotowate (76%) MPNST (21%)
 Duże dłonie i stopy (46%) Skolioza (43%) Nadmierna ruchomość stawów (72%) Opóźnienie rozwoju psychoruchowego, problemy szkolne (93%), zaburzenia koncentracji (73%), opóźnienie rozwoju mowy (48%), NI (38%) Wcześniejsze wystąpienie objawów typowych dla NF-1 (przed 5 rokiem życia) Nerwiakowłókniaki (76%) Nerwiaki splotowate (76%) MPNST (21%)")
38
WRODZONY PRZEROST NADNERCZY
Zaburzenie steroidogenezy, polegające na braku lub obniżonej aktywności enzymów uczestniczących w syntezie kortyzolu lub upośledzeniu jego dostępności do receptora Zwiększone wytwarzanie ACTH Nadmierna produkcja pozostałych hormonów kory nadnerczy (androgeny nadnerczowe i niekiedy mineralokortykoidy) Częstość występowania Postać klasyczna 1:5 tys. – 1:20 tys. żywych urodzeń dla populacji europejskiej (Eskimosi Yupik z Alaski 1:280) Postać nieklasyczna 1:1000 (Polska), 1:300 (Włochy), 1:50 (Hiszpania), 1:27 (Izrael)
 Częstość występowania. Postać klasyczna 1:5 tys. – 1:20 tys. żywych urodzeń dla populacji europejskiej (Eskimosi Yupik z Alaski 1:280) Postać nieklasyczna 1:1000 (Polska), 1:300 (Włochy), 1:50 (Hiszpania), 1:27 (Izrael)")
39
WPN Dziedziczenie:AR Produkt genu: 21-hydroksylaza (21-OH) – niedobór enzymu dotyczy 95% wszystkich przypadków WPN Gen: CYP21A2 Locus genowe: 6p21.33 Rodzaje mutacji: Mutacje punktowe – opisano kilkadziesiąt mutacji, ale 10 z nich odpowiadają za 90% przypadków Delecja części lub całego genu – u 20% chorych Konwersja – przemieszczenie nieaktywnej części pseudogenu do genu aktywnego Korelacja genotyp-fenotyp

40
WPN - MECHANIZM Nadmierna sekrecja ACTH
Przerost kory nadnerczy Nadmierne wydzielanie androgenów nadnerczowych Wirylizacja zewnętrznych narządów płciowych u płodów płci żeńskiej (8-14 tydzień życia płodowego) – obojnactwo rzekome żeńskie U chłopców prawidłowe narządy płciowe – opóźnione rozpoznanie Obniżona czynność rdzenia nadnerczy Dysplazja rdzenia Zmniejszone wytwarzanie adrenaliny – zaburzona odpowiedź na stres, skłonność do nadwagi Hiperinsulinizm Chorzy z WPN są zagrożeni zespołem metabolicznym.
 – obojnactwo rzekome żeńskie. U chłopców prawidłowe narządy płciowe – opóźnione rozpoznanie. Obniżona czynność rdzenia nadnerczy. Dysplazja rdzenia. Zmniejszone wytwarzanie adrenaliny – zaburzona odpowiedź na stres, skłonność do nadwagi. Hiperinsulinizm. Chorzy z WPN są zagrożeni zespołem metabolicznym.")
41
POSTACI WPN Klasyczna – objawy już w okresie życia płodowego
WPN-21OH z utratą soli (SW – salt wasting) Znaczny niedobór kortyzolu i aldosteronu Duże nasilenie objawów Dotyczy 75% przypadków WPN-21OH bez utraty soli (SV – simple virilizing) Synteza aldosteronu nieznacznie ograniczona Łagodniejszy przebieg choroby Mniejsze nasilenie objawów androgenizacji Dotyczy 25% przypadków Nieklasyczna – łagodna, o późnym początku (late onset) zwykle w okresie okołopokwitaniowym Kryptogenna – bez objawów klinicznych, rozpoznawana w oparciu o kryteria biochemiczne i genetyczne
 Znaczny niedobór kortyzolu i aldosteronu. Duże nasilenie objawów. Dotyczy 75% przypadków. WPN-21OH bez utraty soli (SV – simple virilizing) Synteza aldosteronu nieznacznie ograniczona. Łagodniejszy przebieg choroby. Mniejsze nasilenie objawów androgenizacji. Dotyczy 25% przypadków. Nieklasyczna – łagodna, o późnym początku (late onset) zwykle w okresie okołopokwitaniowym. Kryptogenna – bez objawów klinicznych, rozpoznawana w oparciu o kryteria biochemiczne i genetyczne.")
42
OBJAWY – POSTAĆ KLASYCZNA
Z utratą soli Wirylizacja u noworodków żeńskich W 2-3 tygodniu życia (mylone z infekcją przewodu pokarmowego lub zwężeniem odźwiernika): Wymioty, biegunka, odwodnienie Brak łaknienia, słaby przyrost masy ciała Zaburzenia biochemiczne: ↓ glukoza, ↓ Na, ↑ K, ↓ Cl, ↑ mocznik, kwasica metaboliczna Wstrząs Bez utraty soli – dominują objawy androgenizacji Maskulinizacja narządów płciowych u dziewczynek Przyspieszenie wzrastania u obu płci Przedwczesne pokwitanie U chłopców rozpoznanie jest późne – około 4 roku życia.
: Wymioty, biegunka, odwodnienie. Brak łaknienia, słaby przyrost masy ciała. Zaburzenia biochemiczne: ↓ glukoza, ↓ Na, ↑ K, ↓ Cl, ↑ mocznik, kwasica metaboliczna. Wstrząs. Bez utraty soli – dominują objawy androgenizacji. Maskulinizacja narządów płciowych u dziewczynek. Przyspieszenie wzrastania u obu płci. Przedwczesne pokwitanie. U chłopców rozpoznanie jest późne – około 4 roku życia.")
43
OBJAWY – POSTAĆ NIEKLASYCZNA
Pubarche precox Przyspieszone dojrzewanie szkieletowe Przyspieszenie szybkości wzrastania U dziewcząt: Opóźnione menarche Nasilony trądzik Hirsutyzm Męski typ łysienia Wtórny brak miesiączki Objawy zespołu policystycznych jajników U chłopców Wczesne owłosienie na twarzy Nadmierny wzrost prącia w stosunku do wielkości jąder Niski wzrost Oligospermia

44
ROZPOZNANIE Noworodek Łatwe Trudne
Zespół utraty soli (75% wszystkich pacjentów) Płeć żeńska z cechami obojnactwa Trudne Płeć męska bez utraty soli Płeć żeńska bez utraty soli z V stopniem maskulinizacji wg Pradera
 Płeć żeńska z cechami obojnactwa. Trudne. Płeć męska bez utraty soli. Płeć żeńska bez utraty soli z V stopniem maskulinizacji wg Pradera.")
45
BADANIA Badania hormonalne Profil steroidowy moczu (GC/MS) Kariotyp
↑ stężenie 17-OH-progesteronu w surowicy Kortyzol w surowicy może być prawidłowy, ale ↓ rezerwa nadnerczowa (odpowiedź kortyzolu na ACTH) ↑ wydalanie 17-KS w moczu ↑ wydalanie pregnanów w moczu (pregnantriol, prengantriolon) Profil steroidowy moczu (GC/MS) Kariotyp
 ↑ wydalanie 17-KS w moczu. ↑ wydalanie pregnanów w moczu (pregnantriol, prengantriolon) Profil steroidowy moczu (GC/MS) Kariotyp.")
46
BADANIA MOLEKULARNE Aktywność enzymu Fenotyp Mutacja 0% Klasyczny
Delecja całego genu Konwersja Mutacje punktowe: p.Gly111ValfsX21; p.[Ile237Asn; Val238Glu; Met240Lys]; p.Leu308PhefsX6; p.Gln319X; p.Arg357Trp <1% c A>G lub c.293C>G 2-11% p.Ile173Asn 20-50% Nieklasyczny p.Pro31Leu; p.Val282Leu; p.Pro454Ser

47
LECZENIE Przewlekła, systematyczna suplementacja GKS (Hydrokortyzon) i mineralokortykoidów (Cortineff) wg indywidualnego zapotrzebowania pacjenta Zatrzymanie rozwoju maskulinizacji Normalizacja wzrastania Prawidłowy rozwój gonad i zachowanie ich fizjologicznej czynności Rekonstrukcja zewnętrznych narządów płciowych u dziewczynek w 1-2 roku życia, czasem konieczna jest reoperacja w wieku osiągania dojrzałości płciowej Edukacja – samokontrola, systematyczne leczenie, postępowanie w sytuacjach stresowych, prawidłowy rozwój psychoseksualny

48
OPIEKA Porada genetyczna typowa dla dziedziczenia AR
Opieka specjalistyczna: Endokrynolog Psycholog Pomiary auksologiczne Kontrola ciśnienia RTG – wiek kostny

49
POSTĘPOWANIE PRZEDURODZENIOWE
Od 6 Hbd (najpóźniej 7-8 Hbd) leczenie u kobiety ciężarnej – deksametazon w celu zapobieżenia wirylizacji płodu żeńskiego (przechodzi przez łożysko i hamuje produkcję ACTH) Objawy uboczne leczenia: zwiększony przyrost masy ciała, obrzęki, rozstępy Diagnostyka prenatalna Biopsja kosmówki, amniopunkcja Badania hormonalne – 17-OHP Analiza molekularna genu Kariotyp Kontynuacja leczenia hormonalnego w przypadku płodu płci żeńskiej z mutacjami
 leczenie u kobiety ciężarnej – deksametazon w celu zapobieżenia wirylizacji płodu żeńskiego (przechodzi przez łożysko i hamuje produkcję ACTH) Objawy uboczne leczenia: zwiększony przyrost masy ciała, obrzęki, rozstępy. Diagnostyka prenatalna. Biopsja kosmówki, amniopunkcja. Badania hormonalne – 17-OHP. Analiza molekularna genu. Kariotyp. Kontynuacja leczenia hormonalnego w przypadku płodu płci żeńskiej z mutacjami.")
50
INNE BLOKI STEROIDOGENEZY
WPN-11βOH – 5-8% przypadków WPN Objawy: wirylizacja płodów płci żeńskiej, zespół utraty soli oraz nadciśnienie Leczenie – bez mineralokortykoidów WPN-HSD3 – niedobór dehydrogenazy 3β-hydroksysteroidowej Objawy: klasyczne (zespół utraty soli, wirylizacja u dziewczynek, słaba maskulinizacja u chłopców) lub nieklasyczne (pubarche precox, hirsutyzm u kobiet)
 lub nieklasyczne (pubarche precox, hirsutyzm u kobiet)")
51
INNE BLOKI STEROIDOGENEZY
WPN-17αOH – niedobór 17α-hydroksylazy i 17,20-liazy Występuje rzadko – ok.. 1% wszystkich przypadków WPN Objawy: Żeńskie zewnętrzne narządy płciowe u obu płci lub różnego stopnia obojnactwo Hipogonadyzm hipergonadotropowy w okresie dojrzewania Nadciśnienie tętnicze Leczenie: GKS, rekonstrukcja narządów płciowych zewnętrznych i substytucja hormonów płciowych WPN-L – lipoidowy Zaburzenie przezbłonowego transportu cholesterolu do mitochondriów – całkowite upośledzenie steroidogenezy nadnerczowej prowadzące do zgonu Objawy: zawsze żeńskie narządy płciowe (niezależnie od płci), u chłopców konieczne jest usunięcie nieprawidłowych gonad Leczenie: jak w klasycznym WPN z utratą soli oraz substytucja hormonami płciowymi
, u chłopców konieczne jest usunięcie nieprawidłowych gonad. Leczenie: jak w klasycznym WPN z utratą soli oraz substytucja hormonami płciowymi.")
52
ZESPÓŁ CONRADIEGO-HUNERMANNA
Chondrodysplazja punktowa sprzężona z chromosomem X dominująca typu 2 – CDPX2 Dysplazja kostna o zróżnicowanym obrazie klinicznym – od postaci letalnych (dla płodów płci męskiej) do form o niewielkim nasileniu objawów Częstość występowania 1:400 tys., jednak najpewniej jest znacznie wyższa
 do form o niewielkim nasileniu objawów. Częstość występowania 1:400 tys., jednak najpewniej jest znacznie wyższa.")
53
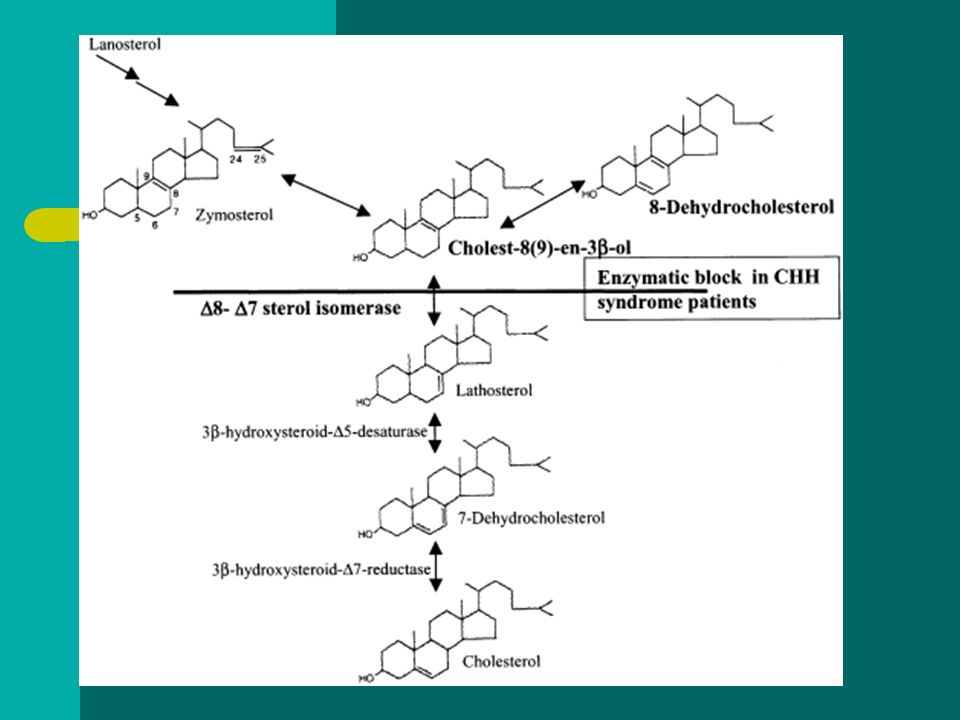
CDPX2 Dziedziczenie:XD Gen: EBP Produkt genu: Emopamil-binding protein
Locus genowe: Xp11.2 Zaburzenie syntezy cholesterolu z powodu niedoboru izomerazy Δ8-Δ7-sterolowej – nadmierne gromadzenie 8-dehydrocholesterolu i cholest-8(9)-en-3β-olu w osoczu i fibroblastach
-en-3β-olu w osoczu i fibroblastach.")
54
OBJAWY Opóźnione wzrastanie i niski wzrost ostateczny Dysmorfia
Wystające czoło Rzadkie brwi i rzęsy (asymetryczne) Płaska nasada nosa Zadarty czubek nosa Wydatne policzki
 Płaska nasada nosa. Zadarty czubek nosa. Wydatne policzki.")
55
OBJAWY Układ kostno-stawowy
Rizomelicze (proksymalne) skrócenie kończyn (często asymetryzcne) Polidaktylia zaosiowa (5%) Przykurcze stawowe Skolioza
 skrócenie kończyn (często asymetryzcne) Polidaktylia zaosiowa (5%) Przykurcze stawowe. Skolioza.")
56
OBJAWY Skóra Erytrodermia lub rybia łuska – charakter rozsiany, obustronnie, wzdłuż linii Blaschko (obszary aktywności chromosomu X z mutacją) Blizny Zaniki skórne o typie orange peel Szorstkie włosy Łysienie Łamliwość paznokci (rzadko)
")
57
OBJAWY Oczy Niedosłuch Rozszczep podniebienia
Zaćma (67%) – wrodzona, asymetryczna Małoocze Niedosłuch Rozszczep podniebienia Wady narządowe: serce, nerki, mózg (tylko u płci męskiej)
 – wrodzona, asymetryczna. Małoocze. Niedosłuch. Rozszczep podniebienia. Wady narządowe: serce, nerki, mózg (tylko u płci męskiej)")
58
BADANIA RTG – punkcikowate ogniska mineralizacji w nasadach kości długich, oraz kręgach, chrząstce tchawicy, dystalnych odcinkach żeber – ustępują przed 12 miesiącem życia Badania biochemiczne Stężenie 8(9)-cholesterolu w surowicy lub fibroblastach - ↑ Stężenie 8-dehydrocholesterolu w surowicy oznaczone metodą GC-MS - ↑ Badania molekularne – sekwencjonowanie (identyfikacja mutacji u 90% pacjentów)
-cholesterolu w surowicy lub fibroblastach - ↑ Stężenie 8-dehydrocholesterolu w surowicy oznaczone metodą GC-MS - ↑ Badania molekularne – sekwencjonowanie (identyfikacja mutacji u 90% pacjentów)")
60
OBJAWY U CHŁOPCÓW Mutacja w genie EBP u pacjenta z zespołem Klinefeltera – cechy typowe Mutacja w genie EBP u pacjenta bez aberracji chromosomowej Dysmorfia: hiperteloryzm, zmarszczka nakątna, wystająca nasada nosa, nisko osadzone uszy, małożuchwie, duże ciemię przednie Zaćma Rybia łuska

61
OBJAWY U CHŁOPCÓW Polidaktylia zaosiowa Syndaktylia 2. i 3. palca stóp
Wnętrostwo, spodziectwo Wady narządowe Serce – VSD, ASD Mózg – hipoplazja móżdżku, agenezja ciała modzelowatego, zespół Dandy’ego-Walkera Głębokie upośledzenie rozwoju psychoruchowego

62
DIAGNOSTYKA RÓŻNICOWA
Rizomeliczna chondrodysplazja punktowa typu 1, 2 i 3 (RCDP1, RCDP2, RCDP3) – zaburzenia biosyntezy peoksysomów Małogłowie Dysmorfia – twarz „misia koala” Zaćma Rizomeliczne skrócenie kończyn górnych, w mniejszym stopniu dolnych Wady kręgów Wady mózgu (opóźniona mielinizacja, wentrikulomegalia, atrofia móżdżku) Padaczka Głębokie upośledzenie wzrastania i rozwoju Dziedziczenie: AR, mutacje w genach (PEX7, DHPAT, AGPS)
 – zaburzenia biosyntezy peoksysomów. Małogłowie. Dysmorfia – twarz „misia koala Zaćma. Rizomeliczne skrócenie kończyn górnych, w mniejszym stopniu dolnych. Wady kręgów. Wady mózgu (opóźniona mielinizacja, wentrikulomegalia, atrofia móżdżku) Padaczka. Głębokie upośledzenie wzrastania i rozwoju. Dziedziczenie: AR, mutacje w genach (PEX7, DHPAT, AGPS)")
63
DIAGNOSTYKA RÓŻNICOWA
CDPX1 (brachytelefalangiczna) Niedorozwój dystalnych paliczków Hipoplazja szczękowo-nosowa – twarz „przyklejona do szyby” Nie stwierdza się skrócenia kończyn, zaćmy Dziedziczenie: XR, mutacja w genie ARSE (Xp)
 Niedorozwój dystalnych paliczków. Hipoplazja szczękowo-nosowa – twarz „przyklejona do szyby Nie stwierdza się skrócenia kończyn, zaćmy. Dziedziczenie: XR, mutacja w genie ARSE (Xp)")
64
DIAGNOSTYKA RÓŻNICOWA
Zaburzenia biosyntezy cholesterolu Zespół CHILD (Congenital Hemidysplasia with Ichthyosiform erytroderma and Limb Defect) Jednostronne, nie przekraczające linii środkowej ciała, ogniska erytrodermii i łysienia Skrócenie lub wady kończyn po tej samej stronie Wady narządowe (serce, mózg, nerki) Dziedziczenie: XD, mutacja genu NSDHL (Xq28) SLOS
 Jednostronne, nie przekraczające linii środkowej ciała, ogniska erytrodermii i łysienia. Skrócenie lub wady kończyn po tej samej stronie. Wady narządowe (serce, mózg, nerki) Dziedziczenie: XD, mutacja genu NSDHL (Xq28) SLOS.")
65
OPIEKA Porada genetyczna – dziedziczenie DX
Opieka wielospecjalistyczna Okulista Laryngolog Ortopeda – zabieg operacyjny? Dermatolog Rehabilitacja

Podobne prezentacje










 – współczynnik powstały przez podzielenie masy.>")








