Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Rudolf Julius Emmanuel Clausius (1822-1888) Ludwig Eduard Boltzmann (1844-1906)
 Ludwig Eduard Boltzmann ( )")
2
Wnioski z I i II zasady (1) dU = -pdV + TdS dU = dw + dQ = dw odw + dQ odw Wnioski: Istnienie związków pomiędzy parametrami (funkcjami) stanu. Uzasadnienie zasady Duhema (dwa parametry opisują różniczkę zupełną). Interpretacja temperatury i możliwe dalsze rozwinięcie dU.
. Interpretacja temperatury i możliwe dalsze rozwinięcie dU..")
3
Wnioski z I i II zasady(2) dU = -pdV + TdS To jest bilans energii: praca +ciepło ! „zwykła” siła ….bo mogą być inne formy przekazywania energii ! parametr intensywny – siła uogólniona deformacja parametru ekstensywnego

4
Wnioski z I i II Zasady (3) dla procesu odwracalnego dla każdego procesu U,V,(N) = const …. entropia rośnie i osiąga maksimum w stanie równowagi (zasada wzrostu entropii)
.")
5
Wnioski z I i II Zasady (4) dla procesu odwracalnego dla każdego procesu S,V,(N) = const …. energia wewnętrzna maleje i osiąga minimum w stanie równowagi

6
Wnioski z I i II Zasady (5) Nie tylko entropia decyduje o naszym Świecie…. Parametrem rozstrzygającym o kierunku zachodzenia procesów mogą być różne funkcje (zwane potencjałami termodynamicznymi). Entropia jest potencjałem termodynamicznym dla U,V, N = const, podczas gdy dla warunków S,V,N = const, takim potencjałem jest energia wewnętrzna. Z praktycznego punktu widzenia najlepszy byłby potencjał „rządzący” procesami w warunkach dających się łatwo kontrolować (stałe parametry p, V, T)
. Entropia jest potencjałem termodynamicznym dla U,V, N = const, podczas gdy dla warunków S,V,N = const, takim potencjałem jest energia wewnętrzna. Z praktycznego punktu widzenia najlepszy byłby potencjał „rządzący procesami w warunkach dających się łatwo kontrolować (stałe parametry p, V, T).")
7
Wnioski z I i II Zasady (6) – pozostałe potencjały dla procesu odwracalnego Entalpia: H = U + pV …. entalpia maleje i osiąga minimum w stanie równowagi dla każdego procesu S,p,(N) = const U = H - pV
 = const U = H - pV.")
8
Wnioski z I i II Zasady (7) – pozostałe potencjały dla procesu odwracalnego Energia swobodna (Helmholtza): F = U - TS …. energia swobodna maleje i osiąga minimum w stanie równowagi dla każdego procesu T,V,(N) = const U = F + TS
 = const U = F + TS.")
9
Wnioski z I i II Zasady (8) – pozostałe potencjały dla procesu odwracalnego Entalpia swobodna (Gibbsa): G = H – TS …. entalpia swobodna maleje i osiąga minimum w stanie równowagi dla każdego procesu T,p,(N) = const U = G – pV + TS = U + pV - TS
 = const U = G – pV + TS = U + pV - TS.")
10
Entalpia swobodna – najważniejszy potencjał termodynamiczny różniczka zupełna Entalpia swobodna (energia Gibbsa, funkcja Gibbsa) G = H – TS pochodne cząstkowe relacja Maxwella
 G = H – TS pochodne cząstkowe relacja Maxwella")
11
Potencjały termodynamiczne – pochodne i różniczki Potencjał termod. różniczka zupełna pochodne cząstkowe relacje Maxwella EntropiadS = (1/T)dU + (p/T)dV ( S/ U) V = 1/T ( S/ V) U = p/T Energia wewnętrzna dU = TdS - pdV ( U/ S) V = T ( U/ V) S = -p ( T/ V) S = - ( p/ S) V EntalpiadH = TdS +Vdp ( H/ S) p = T ( H/ p) S = V ( T/ p) S = ( V/ S) p Energia swobodna dF = -SdT - pdV ( F/ T) V = -S ( F/ V) T = -p ( S/ V) T = ( p/ T) V Entalpia swobodna dG = -SdT +Vdp ( G/ T) p = -S ( G/ p) T = V ( S/ p) T = - ( V/ T) p
dU + (p/T)dV ( S/ U) V = 1/T ( S/ V) U = p/T Energia wewnętrzna dU = TdS - pdV ( U/ S) V = T ( U/ V) S = -p ( T/ V) S = - ( p/ S) V EntalpiadH = TdS +Vdp ( H/ S) p = T ( H/ p) S = V ( T/ p) S = ( V/ S) p Energia swobodna dF = -SdT - pdV ( F/ T) V = -S ( F/ V) T = -p ( S/ V) T = ( p/ T) V Entalpia swobodna dG = -SdT +Vdp ( G/ T) p = -S ( G/ p) T = V ( S/ p) T = - ( V/ T) p.")
12
Potencjały termodynamiczne PotencjałParametryWarunek S (II zasada)U,V(dS) U,V ≥ 0 U (I zasada)S,V(dU) S,V ≤ 0 H = U + pVS, p(dH) S,p ≤ 0 F = U - TST, V(dF) T,V ≤ 0 G = H - TST, p(dG) T,p ≤ 0
U,V(dS) U,V ≥ 0 U (I zasada)S,V(dU) S,V ≤ 0 H = U + pVS, p(dH) S,p ≤ 0 F = U - TST, V(dF) T,V ≤ 0 G = H - TST, p(dG) T,p ≤ 0")
13
Wnioski z I i II Zasady Termodynamiki 1. Istnieją funkcje (potencjały termodynamiczne), których zmiana, przy stałości dwóch parametrów, decyduje o kierunku procesu; potencjał termodynamiczny osiąga minimum (maksimum) w stanie równowagi. 3. Można wyprowadzić liczne tożsamości, wyrażające związki pomiędzy funkcjami termodynamicznymi, umożliwiające obliczanie ich zmian podczas rzeczywistych procesów. 2. Daje to możliwość znajdywania związków między parametrami w stanie równowagi.
, których zmiana, przy stałości dwóch parametrów, decyduje o kierunku procesu; potencjał termodynamiczny osiąga minimum (maksimum) w stanie równowagi. 3. Można wyprowadzić liczne tożsamości, wyrażające związki pomiędzy funkcjami termodynamicznymi, umożliwiające obliczanie ich zmian podczas rzeczywistych procesów. 2. Daje to możliwość znajdywania związków między parametrami w stanie równowagi..")
14
Zależność entropii od temperatury

15
Jak wyznaczyć entropię? cpcp lnT lnT 1 lnT 0

16
III Zasada Termodynamiki Jeśli przyjmiemy, że S(T=0) = 0 - postulat ten nosi nazwę III Zasady Termodynamiki W termodynamice statystycznej wymóg ten jest zbyteczny, bo dla S(Ω =1) = kln(1) = 0 i ten stan odpowiada T = 0
 = 0 - postulat ten nosi nazwę III Zasady Termodynamiki W termodynamice statystycznej wymóg ten jest zbyteczny, bo dla S(Ω =1) = kln(1) = 0 i ten stan odpowiada T = 0")
17
Sformułowanie Plancka (1911) T → 0, S → 0 Sformułowanie Nernsta (1905) T → 0, ∆S → 0 Niektóre konsekwencje: c p → 0 dla T → 0 niemożność osiągnięcia T = 0!
 T → 0, S → 0 Sformułowanie Nernsta (1905) T → 0, ∆S → 0 Niektóre konsekwencje: c p → 0 dla T → 0 niemożność osiągnięcia T = 0!")
18
Termodynamika układów otwartych Bilans energii: dU = – pdV + TdS + …… ? przecież U zmienia się w wyniku transportu masy!!!!

19
Potencjał chemiczny Potencjał chemiczny - ma charakter siły uogólnionej, - jest miarą wpływu zmiany liczby moli na energię wewnętrzną, est parametrem intensywnym

20
Różniczka zupełna energii wewnętrznej

21
Równowaga w układzie wieloskładnikowym i wielofazowym (1) U, V, N = const układ jako całość izolowany, poszczególne fazy nie są izolowane względem siebie; możliwe: przekazywanie energii wewnętrznej (na sposób ciepła), zmiana objętości (praca objętościowa) i przenoszenie masy (dyfuzja składników) U = U + U = const V = V + V = const N i = N i + N i = const αβ W warunkach równowagi entropia całego układu osiąga maksimum! izolacja od otoczenia

22
Równowaga w układzie wieloskładnikowym i wielofazowym (2) U, V, N = const U = U + U V = V + V N i = N i + N i αβ W warunkach równowagi dS = dS α + dS β = 0
 U, V, N = const U = U + U V = V + V N i = N i + N i αβ W warunkach równowagi dS = dS α + dS β = 0")
23
Równowaga w układzie wieloskładnikowym i wielofazowym (3) U, V, N = const U = U + U V = V + V N i = N i + N i αβ Parametrami niezależnymi są tylko te, odnoszące się do jednej fazy - α albo β: dU + dU = 0 dV + dV = 0 dn i + dn i = 0 Konieczność zerowania się pochodnych cząstkowych! Parametrami niezależnymi są tylko te, odnoszące się do jednej fazy - α albo β: dU = - dU dV = - dV dn i = - dn i

24
Równowaga w układzie wieloskładnikowym i wielofazowym (4) p = p = p =... = p T = T = T =... = T i = i = i =... = i (dla każdej fazy) (dla każdego składnika i = 1, 2, 3,...,k)
 (dla każdego składnika i = 1, 2, 3,...,k).")
25
Warunki stabilności – warunki konieczne występowania maksimum entropii warunek stabilności termicznej: c v ≥ 0 warunek stabilności mechanicznej:

26
Reguła faz (Gibbsa) (1) Układ składa się z f faz i n składników liczba parametrów intensywnych = 2 + f(n - 1) [T,p + ułamki molowe dla każdej z faz] Równowaga w układzie wieloskładnikowym i wielofazowym wynika z wartości parametrów intensywnych (T, p, μ i ) i w związku z czym nie zależy od wielkości układu. Parametry intensywne określające stan układu to – T, p, stężenia (a nie liczby moli!) Stężenia mogą być różnie zdefiniowane – np. ułamki molowe – x k = n k /∑n i Dla układu n-składnikowego mamy n-1 niezależnych stężeń liczba parametrów niezależnych (stopni swobody układu - ) = liczba parametrów – liczba równań = 2 + f(n-1) - n(f-1) i = i = i =... = i (dla każdego składnika i = 1, 2, 3,...,k) liczba równań wiążących te parametry = n(f - 1) [równość potencjałów chemicznych] = 2 + nf – f – nf + n = n + 2 - f
![Reguła faz (Gibbsa) (1) Układ składa się z f faz i n składników liczba parametrów intensywnych = 2 + f(n - 1) [T,p + ułamki molowe dla każdej z faz] Równowaga w układzie wieloskładnikowym i wielofazowym wynika z wartości parametrów intensywnych (T, p, μ i ) i w związku z czym nie zależy od wielkości układu.](http://images.slideplayer.pl/38/10764971/slides/slide_26.jpg "Parametry intensywne określające stan układu to – T, p, stężenia (a nie liczby moli!) Stężenia mogą być różnie zdefiniowane – np. ułamki molowe – x k = n k /∑n i Dla układu n-składnikowego mamy n-1 niezależnych stężeń liczba parametrów niezależnych (stopni swobody układu - ) = liczba parametrów – liczba równań = 2 + f(n-1) - n(f-1) i = i = i =... = i (dla każdego składnika i = 1, 2, 3,...,k) liczba równań wiążących te parametry = n(f - 1) [równość potencjałów chemicznych] = 2 + nf – f – nf + n = n f.")
27
Reguła faz (2) = n + 2 - f Przykład 1: Substancja czysta, równowaga ciecz-para = 1 + 2 – 2 = 1 Parametry: T, p Związek pomiędzy parametrami μ c (T,p) = μ g (T,p)
 = n f Przykład 1: Substancja czysta, równowaga ciecz-para = – 2 = 1 Parametry: T, p Związek pomiędzy parametrami μ c (T,p) = μ g (T,p)")
28
Reguła faz (3) = n + 2 - f Przykład 2: Maksymalna liczba faz, które mogą współistnieć w równowadze - f max f = n + 2 - f max = n + 2 - ( min = 0) = n + 2 Dla substancji czystej f max = 3 (punkt potrójny)
 = n f Przykład 2: Maksymalna liczba faz, które mogą współistnieć w równowadze - f max f = n f max = n ( min = 0) = n + 2 Dla substancji czystej f max = 3 (punkt potrójny)")
29
Warunki równowagi - przykład c Jakie równania muszą być spełnione, aby poniższy układ znajdował się w stanie równowagi? g (s) – NaCl (c) – H 2 O + NaCl + aceton (ac) (g) – H 2 O + aceton + powietrze 1. Równość T = (T c = T s = T g ) 2. Równość p = (p c = p s = p g ) 3. μ s N aCl = μ c N aCl 4. μ c H2O = μ g H2O 5. μ c ac = μ g ac s
 – NaCl (c) – H 2 O + NaCl + aceton (ac) (g) – H 2 O + aceton + powietrze 1. Równość T = (T c = T s = T g ) 2. Równość p = (p c = p s = p g ) 3. μ s N aCl = μ c N aCl 4. μ c H2O = μ g H2O 5. μ c ac = μ g ac s.")
30
Josiah Willard Gibbs (1839-1903) 1863Doktorat "On the Form of the Teeth of Wheels in Spur Gearing (O kształcie zębów w przekładniach zębatych) 1866- 1869 Pobyt w Europie 1871-Profesor „fizyki matematycznej” (mathematical physics) w Yale 1875, 1878 On the Equilibrium of Heterogenous Substances 1880Oferta pracy z Johns Hopkins University w Baltimore, przebita przez $ 2000 zaoferowane przez Yale College Prace dotyczące rachunku wektorowego i optyki 1902Elementary Principles in Statistical Mechanics
 1863Doktorat On the Form of the Teeth of Wheels in Spur Gearing (O kształcie zębów w przekładniach zębatych) Pobyt w Europie 1871-Profesor „fizyki matematycznej (mathematical physics) w Yale 1875, 1878 On the Equilibrium of Heterogenous Substances 1880Oferta pracy z Johns Hopkins University w Baltimore, przebita przez $ 2000 zaoferowane przez Yale College Prace dotyczące rachunku wektorowego i optyki 1902Elementary Principles in Statistical Mechanics")
31
Konsekwencje I i II Zasady (1) Termodynamiczne równanie stanu (1) U = F + TS bo F jest potencjałem termodynamicznym dla (T,V) ciśnienie wewnętrzne Termodynamiczne równanie stanu
 Termodynamiczne równanie stanu (1) U = F + TS bo F jest potencjałem termodynamicznym dla (T,V) ciśnienie wewnętrzne Termodynamiczne równanie stanu")
32
Dla gazu doskonałego Konsekwencje I i II Zasady (2) Termodynamiczne równanie stanu (2) Podobnie dla Wniosek – energia wewnętrzna i entalpia gazu doskonałego zależą tylko od temperatury Entalpia dla gazu doskonałego co można wyprowadzić z założeń molekularnych
 Termodynamiczne równanie stanu (2) Podobnie dla Wniosek – energia wewnętrzna i entalpia gazu doskonałego zależą tylko od temperatury Entalpia dla gazu doskonałego co można wyprowadzić z założeń molekularnych")
33
Oddziaływania międzycząsteczkowe – zależność współczynnika kompresji od ciśnienia Z p Z=1 V < V id – dominacja sił przyciągających V > V id – dominacja sił odpychających T=const

34
Współczynnik kompresji http://www.chem.ufl.edu/~itl/2045/lectures/lec_e.html

35
Współczynnik kompresji http://www.chem.ufl.edu/~itl/2045/lectures/lec_e.html N2N2

36
Potencjał oddziaływania dwóch cząsteczek r σ

37
Potencjał Lennarda-Jonesa udział przyciągający udział odpychający

38
Równanie van der Waalsa (1) udział odpychający udział przyciągający
 udział odpychający udział przyciągający")
40
Równanie van der Waalsa (2) Postać matematyczna – równanie sześcienne względem V Możliwe wielokrotne pierwiastki względem V!
 Postać matematyczna – równanie sześcienne względem V Możliwe wielokrotne pierwiastki względem V!")
41
Równanie van der Waalsa (3) Zasada równych pól Maxwella: S 1 = S 2 p V T=const S1S1 S2S2 VcVc VgVg niemożliwe, bo (∂p/∂V) T > 0!!!
 Zasada równych pól Maxwella: S 1 = S 2 p V T=const S1S1 S2S2 VcVc VgVg niemożliwe, bo (∂p/∂V) T > 0!!!")
42
Równanie van der Waalsa (4) Izotermy van der Waalsa dla H 2 O T k = 647,3K p k = 220,5 bar V k = 56 cm 3 /mol punkt krytyczny
 Izotermy van der Waalsa dla H 2 O T k = 647,3K p k = 220,5 bar V k = 56 cm 3 /mol punkt krytyczny")
43
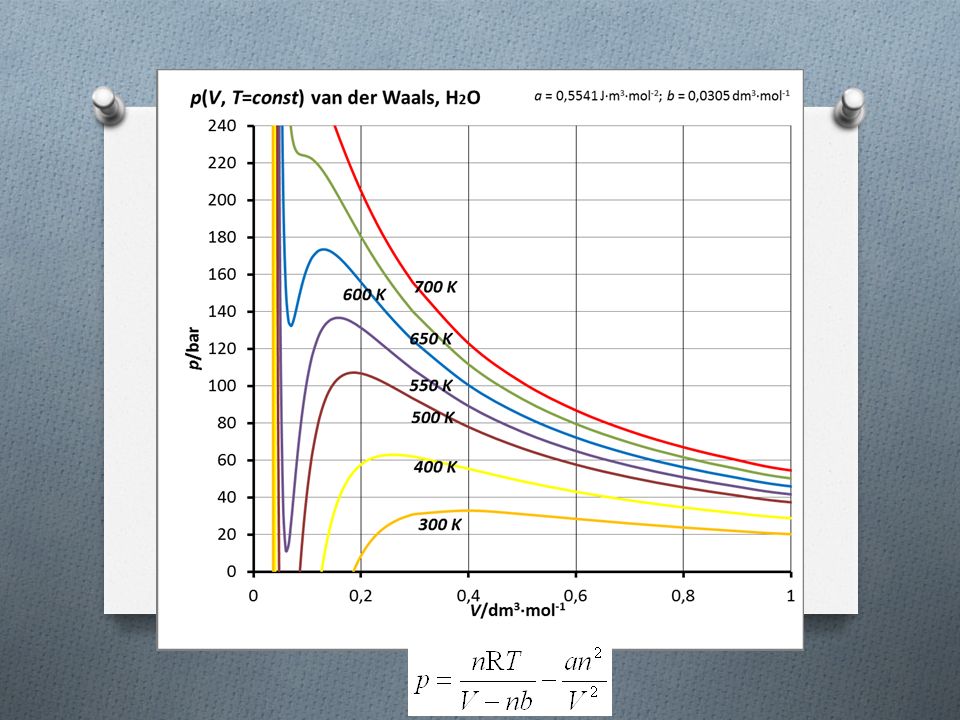
Równanie van der Waalsa (5) Izotermy van der Waalsa dla H 2 O
 Izotermy van der Waalsa dla H 2 O")
44
Równanie van der Waalsa (6) Izotermy van der Waalsa dla H 2 O
 Izotermy van der Waalsa dla H 2 O")
45
równanie van der Waalsa skąd wziąć parametry? Izotermy van der Waalsa dla H 2 O

46
równanie van der Waalsa – parametry z właściwości stanu krytycznego T k = 8a/27Rb p k = a/27b 2 V k = 3b warunek matematyczny punktu krytycznego: parametry van der Waalsa w funkcji parametrów krytycznych: parametry krytyczne w funkcji parametrów van der Waalsa:

47
Wyścig do bieguna zimna Temperatury krytyczne niektórych gazów T k /KT t (p = 1 atm) H2OH2O647,3273,2 NH 3 405,6195,4 C2H4C2H4 282,4104,0 CH 4 190,690,7 NO180106,5 O2O2 154,654,4 CO132,968,0 N2N2 126,263,3 H2H2 33,214,0 He5,3
 H2OH2O647,3273,2 NH 3 405,6195,4 C2H4C2H4 282,4104,0 CH 4 190,690,7 NO180106,5 O2O2 154,654,4 CO132,968,0 N2N2 126,263,3 H2H2 33,214,0 He5,3")
48
Nieco historii kiedycokto 1873równanie van der Waalsa J.D. van der Waals 1883skroplenie powietrzaK. Olszewski 1898skroplenie wodoruJ. Dewar 1908 (10.07, 5:45-19:30) skroplenie heluH. Kamerlingh Onnes 1911nadprzewodnictwo rtęci 1995kondensat Bosego- Einsteina, 10 -9 K E.A. Cornell W. Ketterle C.E. Wieman (Nobel 2001) J.D. van der WaalsK. Olszewski J. DewarH. Kamerlingh Onnes T k /KT t (p = 1 atm) H2OH2O647,3273,2 NH 3 405,6195,4 C2H4C2H4 282,4104,0 CH 4 190,690,7 NO180106,5 O2O2 154,654,4 CO132,968,0 N2N2 126,263,3 H2H2 33,214,0 He5,3
 skroplenie heluH. Kamerlingh Onnes 1911nadprzewodnictwo rtęci 1995kondensat Bosego- Einsteina, K E.A. Cornell W. Ketterle C.E. Wieman (Nobel 2001) J.D. van der WaalsK. Olszewski J. DewarH. Kamerlingh Onnes T k /KT t (p = 1 atm) H2OH2O647,3273,2 NH 3 405,6195,4 C2H4C2H4 282,4104,0 CH 4 190,690,7 NO180106,5 O2O2 154,654,4 CO132,968,0 N2N2 126,263,3 H2H2 33,214,0 He5,3.")
49
Wady równania van der Waalsa Niedokładności w ilościowym opisie stanu krytycznego (błędna wartość Z k = 3/8 = 0,375). równowagi ciecz-para. właściwości cieczy.

50
Błędy równania van der Waalsa Izotermy van der Waalsa dla H 2 O V k = 56 cm 3 /mol

51
Uproszczone formy równania van der Waalsa i co z nich wynika (1) 1. a = 0 (brak oddziaływań przyciągających)
.")
52
Uproszczone formy równania van der Waalsa i co z nich wynika (2) p V T = const
 p V T = const")
53
Uproszczone formy równania van der Waalsa i co z nich wynika (3) 2. b = 0 (brak oddziaływań odpychających)
.")
54
Uproszczone formy równania van der Waalsa i co z nich wynika (4) p T = const p V
 p T = const p V")
55
Uproszczone formy równania van der Waalsa i co z nich wynika (5) Wniosek – występowanie fazy ciekłej jest wspólnym skutkiem istnienia oddziaływań przyciągających i odpychających
 Wniosek – występowanie fazy ciekłej jest wspólnym skutkiem istnienia oddziaływań przyciągających i odpychających")
Podobne prezentacje



 udział odpychający udział przyciągający.>")
U,V(dS) U,V ≥ 0 U (I zasada)S,V(dU) S,V ≤ 0 H = U + pVS, p(dH) S,p ≤ 0 F = U - TST, V(dF)>")

 1. Energia mechaniczna 2. Praca 3.>")


1806 r. - J. Berzelius wprowadził nazwę „związki organiczne” dla wszystkich substancji występujących w organizmach roślinnych.>")




 (1623-1662) Blaise Pascal Ciśnienie wywierane na ciecz rozchodzi się jednakowo we wszystkich.>")



 Cechowanie (PN-EN )>")
