Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Stwardnienie zanikowe boczne ALS* (Amyotrophic lateral sclerosis)
Dawid Szymański *w polskojęzycznej literaturze medycznej i naukowej SLA

2
Nazwy i akronimy w Polsce i na świecie
ALS (amyotrophic lateral sclerosis) ELA (la esclerosis lateral amiotrófica) SLA (sclerosis lateralis amyotrophica) Choroba Charcot – Francja i świat MND (z ang. choroba neuronu ruchowego, tj. Motor Neuron disease) Choroba Lou Gehriga – w USA
 ELA (la esclerosis lateral amiotrófica) SLA (sclerosis lateralis amyotrophica) Choroba Charcot – Francja i świat. MND (z ang. choroba neuronu ruchowego, tj. Motor Neuron disease) Choroba Lou Gehriga – w USA.")
3
Lou Gehrig

4
Sunday, May 2, 1939, will be forever remembered in the annals of baseball as the day New York Yankees' first baseman Lou Gehrig voluntarily benched himself, ending a streak of 2,130 consecutive games. For months the oncegreat player's game had been in decline. His reflexes were off. He stumbled, fumbled, and struggled to hit or catch the ball. No one understood why, least of all Gehrig himself. A few weeks after Gehrig benched himself, doctors diagnosed his illness as amyotrophic lateral sclerosis (ALS), a progressive disease of the central nervous system that remains incurable to this day. Two years later, on June 2, 1941, Gehrig died at the age of 37. The disease that took his life became known to Americans as Lou Gehrig's disease. His consecutive games record stood for 56 years until it was broken by the Baltimore Orioles' Cal Ripken Jr. on Sept. 6, 1995.
, a progressive disease of the central nervous system that remains incurable to this day. Two years later, on June 2, 1941, Gehrig died at the age of 37. The disease that took his life became known to Americans as Lou Gehrig s disease. His consecutive games record stood for 56 years until it was broken by the Baltimore Orioles Cal Ripken Jr. on Sept. 6,")
5
SLA - definicja Pierwotna, postępująca choroba neurodegeneracyjna prowadząca do nieodwracalnego uszkodzenia neuronów ruchowych, kory mózgowej, pnia mózgu, i rogów przednich rdzenia kręgowego, a w konsekwencji do śmierci w ciągu kilku lat. Śmierć następuje zwykle na skutek niewydolności oddechowej.

6
Podstawę tych odmiennych klinicznie zespołów chorobowych stanowi jeden ten sam proces patofizjologiczny Podobne zmiany histopatologiczne wystepują we wszystkich odmianach klinicznych, a po początkowych objawach ze strony jednego neuronu ruchowego dołączają się zwykle obiawy z neuronu drugiego

11
Neuroanatomia Droga korowo-rdzeniowa boczna(piramidowa)
Całkowita mielinizacja jej włókien nastepuje w 2r.ż. Odpowiada za dowolne, precyzyjne ruchy, zwłaszcza kończyn Rozpoczyna się w komórkach nerwowych V warstwy kory mózgowej(pola 6 i 4) a kończy na neuronach ruchowych rogu przedniego rdzenia Droga ta ma ok włókien a średnica poszczególnego włókna to ok. 1 do 4 mikrometrów Włókna tej drogi są w 67% zmielinizowane Przebieg : przechodzi przez odnoge tylną torebki wewnetrznej, przez śrofdkowe 3/5 odnogi mózgu, podstawe mostu, tworzy piramidy rdzenia przedłużonego, 90% włókien krzyżuje się w dolnej cześci rdzenia przedłużonego(skrzyzowanie piramid), biegnie w tylnej części(tylnym kwadrancie) sznura bocznego rdzenia kregowego Droga korowo-rdzeniowa przednia Jest to niewielka droga nieskrzyżowana, która biegnie do odpowiedniego segmentu rdzenia i przechodzi przez spoidło białe na stronę przeciwna Jest zwiazana z kontrolą mięśni osiowych(mieśni tułowia) Odgrywa role w kontrolowaniu napiecia zginaczy Biegnie w sznurze przednim Droga przedsionkowo-rdzeniowa Rozpoczyna się w jądrze przedsionkowym bocznym po tej samej stronie Odgrywa role w kontrolowaniu napiecia prostowników Droga czerwienno-rdzeniowa Rozpoczyna się w przeciwległym jądrze czewinnym(śródmózgowie) Droga korowo-opuszkowa , od komórek nerwowych kory mózgowej do jader nerów ruchówych poza nerwami III, IV i VI
 a kończy na neuronach ruchowych rogu przedniego rdzenia. Droga ta ma ok włókien a średnica poszczególnego włókna to ok. 1 do 4 mikrometrów. Włókna tej drogi są w 67% zmielinizowane. Przebieg : przechodzi przez odnoge tylną torebki wewnetrznej, przez śrofdkowe 3/5 odnogi mózgu, podstawe mostu, tworzy piramidy rdzenia przedłużonego, 90% włókien krzyżuje się w dolnej cześci rdzenia przedłużonego(skrzyzowanie piramid), biegnie w tylnej części(tylnym kwadrancie) sznura bocznego rdzenia kregowego. Droga korowo-rdzeniowa przednia. Jest to niewielka droga nieskrzyżowana, która biegnie do odpowiedniego segmentu rdzenia i przechodzi przez spoidło białe na stronę przeciwna. Jest zwiazana z kontrolą mięśni osiowych(mieśni tułowia) Odgrywa role w kontrolowaniu napiecia zginaczy. Biegnie w sznurze przednim. Droga przedsionkowo-rdzeniowa. Rozpoczyna się w jądrze przedsionkowym bocznym po tej samej stronie. Odgrywa role w kontrolowaniu napiecia prostowników. Droga czerwienno-rdzeniowa. Rozpoczyna się w przeciwległym jądrze czewinnym(śródmózgowie) Droga korowo-opuszkowa , od komórek nerwowych kory mózgowej do jader nerów ruchówych poza nerwami III, IV i VI.")
12
Neuroanatomia Ośrodkowy neuron ruchowy
Są to neurony korowe, które daja poczatek drogom korowo-opuszkowym i korowo-rdzeniowym Występują również w jądrach pnia mózgu, które wypływają na obwodowe neurony ruchowe (np. jądro przedsionkowe boczne i jądro czerwienne) Dochodzą do obwodowych neuronów ruchowych, bezpośrednio lub przez interneurony
 Dochodzą do obwodowych neuronów ruchowych, bezpośrednio lub przez interneurony.")
13
Neuroanatomia Cechy uszkodzenia ośrodkowego neuronu ruchowego
Niedowład spastyczny Wzmozone napiecie mięśniowe Zmiejszenie lub zniesienie powierzchownych (skórnych) odruchów : brzusznego i nosidłowego(mosznowego) klonus
 odruchów : brzusznego i nosidłowego(mosznowego) klonus.")
14
Neuroanatomia Obwodowe neurony ruchowe
Są to neurony , które bezpośrednio unerwiają mięśnie szkieletowe Znajdują się one w rogach przednich rdzenia kręgowego Znajdują się one w jądrach ruchowych nerwów III-VII i IX-XII

15
Neuroanatomia Cechy uszkodzenia neuronu obwodowego : Porażenie wiotkie
Brak odruchów(arefleksja) Zanik mieśni(atrofia) Drżenie włókienkowe i pęczkowe mięśni
 Zanik mieśni(atrofia) Drżenie włókienkowe i pęczkowe mięśni.")
19
Patomorfologia ALS W badaniu makroskopowym :
Przednie korzenie rdzenia kręgowego są cienkie, lub zanikłe (w zgrubieniach rdzenia) W bardzo ciężkich przypadkach zanik kory ruchowej i/lub przedruchowej wykazany w sekcji mózgu Sekcja zwłok ujawnia zaniki mięśni, niekiedy bardzo rozległy i nasilony
 W bardzo ciężkich przypadkach zanik kory ruchowej i/lub przedruchowej wykazany w sekcji mózgu. Sekcja zwłok ujawnia zaniki mięśni, niekiedy bardzo rozległy i nasilony.")
20
Hipotezy ENDOGENNE : Genetyczna : 5-10% przypadków ALS to przypadki rodzinne. Wiek zachorowania jest tu wczesny, pierwsze objawy dotyczą zwykle kończyn dolnych. Istnieją przypuszczenia co do dziedziczenia autosomalnego zarówno recesywnego jak i dominującego, chromosom 21 i sąsiedztwo 1. receptora dla aminokwasu pobudzającego kw. Kainowego, 2. Cu/Zn nadtlenkowa dysmutaza – wymiatacz wolnych rodników, hipoteza steresu Defektu w syntezie DNA : defekt te ma prowadzić od syntezy odbiegającego od normy białka w komórkach nerwowych i ich przedwczesnej śmierci. Potwierdzeniem tej hipotezy ma być stwierdzony w niektórych badaniach spadek cytoplazmatycznego RNA oraz uszkodzenie „naprawczego” mechanizmu DNA w komórkach limfoidalnych i fibroblastach chorego Endogennych czynników cytotoksycznych : oparta jest na obserwacji cytotoksycznego wpływu surowicy chorego na ALS na hodowle rdzenia noworodków mysich oraz na krwinki czerwone. Hipoteza jest problematyczna, ponieważ nie stwierdzono podobnej korelacji podczas dootrzewnowego wstrzykniecia u myszy, ani na innych hodowlach

21
Hipotezy ENDOGENNE : O roli receptorów i przekazników : oparta jest na stwierdzeniu spadku receptorów muskarynowych, glicynowych i benzodiazepinowych O roli nowotworów : oparta jest na stwierdzeniu grupy chorych ze współistnieniem choroby neuronu ruchowego(Motor Neuron Disease –MND) oraz raka różnych narządów O roli ubytku heksozaminidazy A Dotyczaca wpływu inwolucji : oparta jest na stwierdzeniu w ALS szeregu zmian histopatologicznych wystepujących w procesie starzenia(zanik i ubytek komórek, spadek zdolności regeneracyjnych, spadek syntezy RNA i nagromadzenie lipofuscyny w komórkach nerwowych)
 oraz raka różnych narządów. O roli ubytku heksozaminidazy A. Dotyczaca wpływu inwolucji : oparta jest na stwierdzeniu w ALS szeregu zmian histopatologicznych wystepujących w procesie starzenia(zanik i ubytek komórek, spadek zdolności regeneracyjnych, spadek syntezy RNA i nagromadzenie lipofuscyny w komórkach nerwowych)")
22
Hipotezy Immunologiczna : opiera ise na wykazaniu u niektórych chorych : Odkładnia się IgG i kompleksów C3 w kłębuszkach nerkowych oraz w korze ruchowej i rdzeniu Zwiazku z patologią układu komórek plazmatycznych Zróznicowania HLA w przebiegu choroby Współistniejacych chorób autoimmunologicznych Zwiazek z paraproteinami IgG, IgA, IgM, które niekiedy przebiegaja pod postacią radiukuloneuropatii i rdzeniowego zaniku mięśni Wzrostu miana przeciwciał przeciwko gangliozydom GM1 Zaburzeń transportu aksonalnego : opiera się na obecności sferoidów w pobliżu komórki nerwowej, lub w dalszej cześci wypustki aksonalnej. W badaniu ulstrastrukturalnym odpowiadaja zageszczeniom neurofilamentów. Wyniki obserwacji potwierdzają hipoteze, lecz brak jest wiedzy co do ewentualnych pierwotnych przyczyn Braku czynnika troficznego(beta-NGF) : brak powierdzenia czynnika antagonistycznego względem beta-NGF dla neuronów ruchowych The familial form of ALS is generally inherited in an autosomal dominant fashion; in approximately 20% of these patients, there is a mutation in the copper/zinc superoxide dismutase gene (SOD-1). Emerging evidence suggests that the buildup of free radicals plays an important role in pathogenesis. Administration of vitamin E delayed the onset of ALS in the transgenic mouse model, suggesting that oxidative stress may be important in disease initiation. Antioxidants may be helpful for patients, but more studies are needed to confirm their role. Pierwotnego uszkodzenia komórek ruchowych kory (Eisen i wsp. 1992)-
 : brak powierdzenia czynnika antagonistycznego względem beta-NGF dla neuronów ruchowych. The familial form of ALS is generally inherited in an autosomal dominant fashion; in approximately 20% of these patients, there is a mutation in the copper/zinc superoxide dismutase gene (SOD-1). Emerging evidence suggests that the buildup of free radicals plays an important role in pathogenesis. Administration of vitamin E delayed the onset of ALS in the transgenic mouse model, suggesting that oxidative stress may be important in disease initiation. Antioxidants may be helpful for patients, but more studies are needed to confirm their role. Pierwotnego uszkodzenia komórek ruchowych kory (Eisen i wsp. 1992)-")
23
Hipotezy ENDOGENNE : Defektu błony : opiera się na stwierdzeniu defektu błony w erytrocytach u chorych na ALS i supozycji podobnego defektu błony w komórkach nerwowyh. Nie znaleziono zaburzen fizyko-chemicznych w błonie O działaniu androgenów : oparta jest na stwierdzeniu duzej liczby receptorów androgenów wkomórkach, które u człowieka z ALS objete są procesem chorobowym O związku z chorobami tarczycy : opera się na stwierdzeniu nadczynności tarczycy u chorych na ALS. Nie została potwierdzona na wiekszym materiale O zwiazku z metabolizmem wapnia : oparta jest na istnieniu klinicznych, nerwowo-mięśniowych przejawów nadczynności przytarczyc. Nie znalazła wystarczającego potwierdzenia O związku z zaburzeniami węglowodanów : oparta jest na roli hipoglikemii w uszkodzeniu komórek nerwowych ,w tym neuronów ruchowych rogu przedniego i ma insulinooporności u chorych na ALS

24
Hipotezy EGZOGENNE : Egzogennych czynników cytotoksycznych : na wyspach Zachodniego Pacyfiku małpy odzywiające się roślinami z rodziny „cykadowej” , zawierającej aminokwac cytotoksyczny(beta-N-metyl-amino-1-alanina) wykazuja zespół objawów klinicznych, zblizony do objawów ALS, (co ciekawe) obserwowanych na tych wyspach. Postuluje się możliwość genetycznych predyspozycji do zmian w metablizmie i wrazliwości na czynniki toksyczne Roli metali cieżkich : zatrucie metalami cieżkimi jak ołów, rteć, mangan imituje zespół objawów klinicznych ALS Wirusowa : przeprowadzono liczne badania z przeszczepianiem tkanki nerwowej zwierzętom doświadczalnym. Stwierdzano zespoły klinicznych objawów imitujących ALS. Obserwowano również kleszczowe zapalenia mózgu i rdzenia imitujące ALS. Próbowano połączyć również post-poli z ALS, jednak pomimo nielicznych, ale zbliżonych objawów klinicznych nie udało się wyizolować wirusa
 wykazuja zespół objawów klinicznych, zblizony do objawów ALS, (co ciekawe) obserwowanych na tych wyspach. Postuluje się możliwość genetycznych predyspozycji do zmian w metablizmie i wrazliwości na czynniki toksyczne. Roli metali cieżkich : zatrucie metalami cieżkimi jak ołów, rteć, mangan imituje zespół objawów klinicznych ALS. Wirusowa : przeprowadzono liczne badania z przeszczepianiem tkanki nerwowej zwierzętom doświadczalnym. Stwierdzano zespoły klinicznych objawów imitujących ALS. Obserwowano również kleszczowe zapalenia mózgu i rdzenia imitujące ALS. Próbowano połączyć również post-poli z ALS, jednak pomimo nielicznych, ale zbliżonych objawów klinicznych nie udało się wyizolować wirusa.")
26
Patomorfologia ALS W badaniu mikroskopowym :
Redukcja w stopniu zależnym od zaawansowania choroby neuronów zlokalizowanych w przednim rogu rdzenia kręgowego na całym jego przebiegu z obserwowalna gliozą(glej gwiazdzisty) i zanikiem mielinizacji włókien nerwowych w przednich korzeniach Podobne zmiany da się zaobserwować w nadrach nerwów czaszkowych : podjezykowym, dwuznacznym i trójdzielnym Pozostałych neuronach okolicy obserwowanej często zawierają ciałka Bunina(PAS-dodatnie cytoplazmatyczne wtręty, które najprawdopodobniej są resztkami autofagocytujących wakuoli komórkowych) Mieśnie wykazują cechy atrofie neurogennej Ubytek komórek w obrebie kory mózgowej występuje w postaci rozlanego przeżedzenia komórkowego, lub też opustoszeń komórkowych, obejmujących jedna lub więcej warst kory(szczególnie często wartwa III i V komórek piramidowych)
 i zanikiem mielinizacji włókien nerwowych w przednich korzeniach. Podobne zmiany da się zaobserwować w nadrach nerwów czaszkowych : podjezykowym, dwuznacznym i trójdzielnym. Pozostałych neuronach okolicy obserwowanej często zawierają ciałka Bunina(PAS-dodatnie cytoplazmatyczne wtręty, które najprawdopodobniej są resztkami autofagocytujących wakuoli komórkowych) Mieśnie wykazują cechy atrofie neurogennej. Ubytek komórek w obrebie kory mózgowej występuje w postaci rozlanego przeżedzenia komórkowego, lub też opustoszeń komórkowych, obejmujących jedna lub więcej warst kory(szczególnie często wartwa III i V komórek piramidowych)")
27
Patomorfologia ALS W badaniu mikroskopowym :
W obrebie pnia mózgu zanik i ubytek komórek jest najwybitniejszy w jadrze nerwu XII oraz w jądrze dwuznacznym U zarłych ludzi w badaniu sekcyjnym stwierdzono zaawansowane zmiany w jadrach nerwów gałkoruchowych, czego nie dało się praktycznie zaobserwować w badaniu przyżyciowym W rdzeniu kregowym najwybitniejsze zaniki obserwuje się w zgrubieniu szyjnym i lędzwiowym W niektórych przypadkach w rogach przednich można znalezć sferoidy ,przejaw odcinkowego rozdęcia akconu(Osm – kule sregrnochłonne, ultrastrukturalne – neurofilamenty) Demielinizacja szlaków korowo-rdzeniowych bocznych i brzusznych giną komórki i wypustki aksonalne, proces zwyrodnieniowy nie przekracza zwykle torebki wewnetrznej Obwodowy układ nerwowy. Zwyrodnienie komórek ruchowych pociąga za soba zwyrodnienie aksonalne we włóknach kożeni przednich oraz włóknach ruchowych nerwów obwodowych Zwyrodnienie aksonalne ruchowe włókna ruchowego prowadzi do zaniku komórek mieśni szkieletowych oraz mieśnia przepony Wśród peczków włókien mieśniowych prawidłowych na poczatku są widoczne peczki włókien zanikłych. Dochodzi tez do nieielkiego stopnia proliferacji tkanki łącznej
 Demielinizacja szlaków korowo-rdzeniowych bocznych i brzusznych giną komórki i wypustki aksonalne, proces zwyrodnieniowy nie przekracza zwykle torebki wewnetrznej. Obwodowy układ nerwowy. Zwyrodnienie komórek ruchowych pociąga za soba zwyrodnienie aksonalne we włóknach kożeni przednich oraz włóknach ruchowych nerwów obwodowych. Zwyrodnienie aksonalne ruchowe włókna ruchowego prowadzi do zaniku komórek mieśni szkieletowych oraz mieśnia przepony. Wśród peczków włókien mieśniowych prawidłowych na poczatku są widoczne peczki włókien zanikłych. Dochodzi tez do nieielkiego stopnia proliferacji tkanki łącznej.")
28
Poprzeczny przekrój przez rdzeń kręgowy obrazujący zanik zmielinizowanych włókien dróg korowo-rdzeniowych

29
Sferoid w badaniu ultrastrukturalnym – nagromadzenie neurofilamentów
Przerost astrocytów w rdzeniu kręgowym i i ch zmiany wsteczne klazmatodendroza (GFAP)
")
30
Słaba barwliwość mieliny w obrębie drogi korowo-rdzeniowej bocznej, wynikająca ze zwyrodnienia aksonalnego(MBP) Wczesne stadium zwyrodnienia aksonalnego w „czesanym” włóknie nerwowym(Osm)
")
31
Sferoidy w „czesanym” włóknie nerwowym(Osm)
Zanik poszczególnych pęczków włókien mięśniowych(HE)
")
32
Neuronofagia(HE) Odcinek L3 rdzenia kręgowego. Zwyrodnienie i cienie komórek ruchowych; w jednej z komórek widoczna wodniczka. Liczne przerosłe astrocyty(HE)
")
33
Ubytki komórkowe i rozrzedzenie podłoza w rogu przednim rdzenia (HE)
Sferoid w poblizu wyrodniejącej komórki rogu przedniego rdzenia (HE)
")
34
Podłużny przekrój sferoidów w częściach aksonu oddalonych od komórki(Gomori)
Kule srebrnochłonne w impregnacji srebrowej w rdzeniu (Gomori)
")
35
Opustoszenia komórkowe w III warstwie kory czołowej
Neuronofagia wyrodniejących komórek nerwowych

36
Częśc jądra nerwu XII w przypadku kontrolnym(45)
Częśc jądra nerwu XII w przypadku kontrolnym(45). Obecne liczne motoneurony Cześć jądra nerwu XII w ALS. Obecne pojedyńcze zwyrodniałe motoneurony oraz sferoid(strzałka)
. Obecne liczne motoneurony. Cześć jądra nerwu XII w ALS. Obecne pojedyńcze zwyrodniałe motoneurony oraz sferoid(strzałka)")
37
Część rogu przedniego C8 w przypadku kontrolnym
Część rogu przedniego C8 w przypadku kontrolnym. Widoczne liczne komórki nerwowe Cześc rogu przedniego C8 w ALS. Wybitny ubytek komórek nerwowych. Widoczne liczne jadra astrocytów

38
Glejoza asrocytarna ,szczególnie nasilona na obrzeżu rogu przedniego

39
Epidemiologia Wystepuje mniej więcej równomiernie na całym świecie z czestotliwością 0,7-1,5/100,000, w Europie najcześciej choruja Szwedzi Najczęściej występuje w srednim wieku IV aż do początku VII dekady życia Jednak znane są przypadki występowania choroby u dużo młodszych, jak i u ludzi w podeszłym wieku Częściej chorują mężczyzni(M>K) jednak nie stwierdzono tej przewagi w przypadku rodzinnego ALS Choroba zwykle trwa 2-3lata, ale stwierdzono też przypadki o krótszym, kilkumiesięcznym przebiegu jaki i o długotrwałym 10 letnim Różne są dane dotyczące wieku zachorowania i przebiegu choroby w przypadkach rodzinnych Przypadki rodzinne stanowią ok. 10% Występowanie choroby nie jest zalezne od rasy, czasu, ani lokalizacji geograficznej More than 30,000 Americans have ALS, according to the ALS Association, a nonprofit organization that supports ALS research and public and patient education about the disease. Around 3,000 to 5,000 new cases of the disease are diagnosed every year.
 jednak nie stwierdzono tej przewagi w przypadku rodzinnego ALS. Choroba zwykle trwa 2-3lata, ale stwierdzono też przypadki o krótszym, kilkumiesięcznym przebiegu jaki i o długotrwałym 10 letnim. Różne są dane dotyczące wieku zachorowania i przebiegu choroby w przypadkach rodzinnych. Przypadki rodzinne stanowią ok. 10% Występowanie choroby nie jest zalezne od rasy, czasu, ani lokalizacji geograficznej. More than 30,000 Americans have ALS, according to the ALS Association, a nonprofit organization that supports ALS research and public and patient education about the disease. Around 3,000 to 5,000 new cases of the disease are diagnosed every year.")
40
Podział wszystkich tych odmian klinicznych wynika z uwarunkowań historycznych
1847 – Duchenne opublikował przypadek wybiórczego uszkodzenia dolnego neuronu ruchowego oraz reakcje mięśni na działanie prądu galwanicznego 1950 – Aran dokonał przeglądu opisanych poprzednio podobnych przypadków i nazwał je postępującym zanikiem mięśni 1860 – Duchanne opisał postepujące porażenie opuszki 1869 – Charcot i Joffroy na podstawie obserwacji przypadków opublkowali klasyczny opis choroby – stwardnienie zanikowe boczne – o przebiegu szybszym niż postepujący zanik mieśni typu Arana-Duchenne’a i charakteryzujacy się objawami uszkodzenia zarówno górnego jak i dolnego neuronu ruchowego Początkowo opisane wyżej zespoły były traktowane jako odrebne, nozologiczne jednoski chorobowe. Dopiero Dejerine znalazł w tych zespołach podobne zmiany morfologiczne, wyspępujące w istocie szarej i biłałej rdzenia kregowego. Zaliczył więc wszystkiej zespoły do stwardnienia zanikowego bocznego.

41
Do wyróżnionych w obecnej chwili odmiennych klinicznych postaci
Obraz kliniczny Do wyróżnionych w obecnej chwili odmiennych klinicznych postaci ALS zaliczają się : Zespół zasadniczy – stwardnienie zanikowe boczne Pierwotne stwardnienie boczne Postępujące porażenie opuszki Zespół imitujacy rdzeniowy zanik mięśni

42
Do wyróżnionych w obecnej chwili odmiennych klinicznych postaci
Obraz kliniczny Do wyróżnionych w obecnej chwili odmiennych klinicznych postaci ALS zaliczają się : Zespół zasadniczy – stwardnienie zanikowe boczne Pierwotne stwardnienie boczne Postępujące porażenie opuszki Zespół imitujacy rdzeniowy zanik mięśni

43
Podstawę tych odmiennych klinicznie zespołów chorobowych stanowi jeden ten sam proces patofizjologiczny Podobne zmiany histopatologiczne wystepują we wszystkich odmianach klinicznych, a po początkowych objawach ze strony jednego neuronu ruchowego dołączają się zwykle obiawy z neuronu drugiego

44
Obraz kliniczny zespółu imitujacego rdzeniowy zanik mięśni
Jeżeli choroba zaczyna się od uszkodzenia motoneuronów rogu przedniego rdzenia kregowego , obraz kliniczny imituje rdzeniowy zanik mięśni Zanik pojawia się najwcześniej w obrebie kłębu kciuka i kłębika nasępnie w pozostałych drobnych mięśniach dłoni Obejmuje mieśnie obręczy barkowej, często symetrycznie, lub tez mięśnie przedramion i ramienia W zanikajacych mięśniach występuje drżenie pęczkowe, również w mięśniach w których zanik nie jest jeszcze widoczny, chorzy odczuwają drżenie pęczkowe jako łaskotanie, „chodzenie robaków pod skórą” W miarę postępu choroby nasila się niedowład zanikjących mięśni, pojawiający się najwcześniej w częściach odsiebnych i doprowadzający do „szpoowatego” lub „małpiego” ustawienia palców Odruchy okostnowo-ściegniste słabną i zanikają Niedowład lub porażenie obejmuje stopniowo całe konczyny górne Analogiczna stuacja ma miejsce w konczynach dolnych(rozległy zanik mięśni, drzenie peczkowe, nasilający się obwodowy niedowład, porazenie czterech konczyn, z wiotkim napięciem mieśni i brakiem odruchów) ostatecznie doprowadzając do całowitego unieruchomienia chorego i jego całkowetej zalezności od pomocy osób trzecixh Taki przebieg wystepuje wtedy gdy proces chorobowy zaczyna się w odcinku szyjnym rdzenia kregowego i rozprzestrzenia się w kierunku doogonowym
 ostatecznie doprowadzając do całowitego unieruchomienia chorego i jego całkowetej zalezności od pomocy osób trzecixh. Taki przebieg wystepuje wtedy gdy proces chorobowy zaczyna się w odcinku szyjnym rdzenia kregowego i rozprzestrzenia się w kierunku doogonowym.")
45
Zanik mięśni międzykostnych dłoni

46
Obraz kliniczny pierwotnego stwardnienia bocznego
Proces chorobowy obejmuje przede wszystkim komórki ruchowe kory mózgu i drobi piramidowe Narastajacy niedowład kurczowy czterech kończyn, zwkle choc nie zawsze symetryczny przejawiajacy się całkowitym niedowładem konczyn, wzmożeniem napiecia mięśniowego, wygórowaniem odruchów fizjologicznych, stopkotrzasem, rzepkotrząsem i objawami patologicznymi oraz brakiem powierzchownych odruchów brzusznych Często nie obserwuje się osrodkowego niedowładu n.VII i XII Nie wystepuje zwykle zaburzenie zwieracza pęcherza moczowego i odbytu Wraz z postepem choroby upadabnia się ona do ALS W przypadku braku zajęcia nauronu dolnego ruchowego należy rozważyć inna przyczyne choroby OUN

47
Obraz kliniczny postępujacego porażenia opuszki
Rozpoczyna się od uszkodzenia rdzenia przedłużonego i postepujacych zaburzeń opuszkowych(zespół opuszkowy i rzekomoopuszkowy) Mowa staje się niewyrazna, pojawiaja się zaburzenia połykania, krztuszenie się Można zwykle stwierdzic : drżenie włókienkowe w obrębie języka, jego zbaczanie, upośledzenie ruchów podniebienia miekkiego i języka, czasem niedowład obwodowy n.VII Niedowład osrodkowy n.VII, żywy odruch żwaczowy oraz smiech i płacz przymusowy wskazuja na zajecie układu piramidowego w procesie chorobowym Bełkotliwa dyzartryczna mowa, w miare postepu dochodzi do niemożności mówienia, jedzenia, łykania Objawy istotne diagnostycznie : Fascykujacje i zanik mięśni języka Wygórowany odruch żuchwowy Osłabienie mięsni zginaczy szyi Zywe odruchy scięgniste mimo zaniku mięśni, fascykulacje w zajętych mieśniach Brak zaburzen czucia
 Mowa staje się niewyrazna, pojawiaja się zaburzenia połykania, krztuszenie się. Można zwykle stwierdzic : drżenie włókienkowe w obrębie języka, jego zbaczanie, upośledzenie ruchów podniebienia miekkiego i języka, czasem niedowład obwodowy n.VII. Niedowład osrodkowy n.VII, żywy odruch żwaczowy oraz smiech i płacz przymusowy wskazuja na zajecie układu piramidowego w procesie chorobowym. Bełkotliwa dyzartryczna mowa, w miare postepu dochodzi do niemożności mówienia, jedzenia, łykania. Objawy istotne diagnostycznie : Fascykujacje i zanik mięśni języka. Wygórowany odruch żuchwowy. Osłabienie mięsni zginaczy szyi. Zywe odruchy scięgniste mimo zaniku mięśni, fascykulacje w zajętych mieśniach. Brak zaburzen czucia.")
48
Obraz kliniczny zespółu imitujacego rdzeniowy zanik mięśni
Proses chorobowy może jednak manifestowac się poczatkowo w odsiebnych cześciach konczyn dolnych – zespół kliniczny może wiec odpowiadać niedowładowi nerwów piszczelowych z lokalizacją procesu chorobowego w odcinku ledzwiowo-krzyżowym rdzenia Na wysokości C2/C4 zlokalizowane jest jądro nerwu przeponowego. Zajecie procesem chorobowym tej lokalizacji powoduje porażenie przepony z objawami niewydolności oddechowej i w kńcu porażenie oddechu W postaci imitującej rdzeniowy zanik mięśni często nieudaje się wykazać klinicznie cech uszkodzenia górnego neuronu ruchowego, nawet objawu Babińskiego

49
Obraz kliniczny postaci klasycznej stwardnienia zanikowego bocznego
Zwykle jednoczasowe zajęcie górnego i dolnego neuronu ruchowego Jeżeli proces chorobowy jest umiejscowiony w odcinku szyjnym rdzenia kregowego to wystepująklasyczne objawy stwardnienia zanikowego bocznego : osłabienie, nie zawsze symetryczne, dłoni i palców, „chodzenie robaków pod skórą”, uczucie sztywnosci nóg i problemy w chodzeniu Poczatkowe stadium ALS charakteryzuje się klinicznie objawami : zanik odsiebnych mieśni kończyn górnych, drzenie peczkowe na tle jeszcze wzmożonego napięcia mieśniowego, i wygórowanych odruchów okostnowo-ścięgnistych, dodatni objaw(odruch) Jacobsohona(polega na zgięciu palców, obustronny jest fizjologiczny, a jednostronny patologiczny), brak odrruchów podeszwowych, lub obecny objaw Babińskiego W miare postępu choroby dołączaja się objawy swiadczące o umiejscowieniu procesu chorobowego w odcinku piersiowym, lędzwiowo-rzyzowym oraz w opuszce : pojawiają się i narastają zaniki mięśni i niedowład kończyn dolnych, rozległe drżenia pęczkowe, drzenia włókienkowe i dyzartria niedowład lub porażenie mięśnia przepony z niedydolnością oddechową przejawiajacą się przyspieszonym płytkim oddechem z udziałem skrzydełek nosa i pomocniczych mięśni oddechowych, tachykardią, szarym zabarwieniem powłok i niepokojem w zaawansowanej postaci ALS z zespołem opuszkowym i rzekomoopuszkowym bardzo precyzyjne badania ruchów gałek ocznych wykazują ich zaburzenia
 Jacobsohona(polega na zgięciu palców, obustronny jest fizjologiczny, a jednostronny patologiczny), brak odrruchów podeszwowych, lub obecny objaw Babińskiego. W miare postępu choroby dołączaja się objawy swiadczące o umiejscowieniu procesu chorobowego w odcinku piersiowym, lędzwiowo-rzyzowym oraz w opuszce : pojawiają się i narastają zaniki mięśni i niedowład kończyn dolnych, rozległe drżenia pęczkowe, drzenia włókienkowe i dyzartria. niedowład lub porażenie mięśnia przepony z niedydolnością oddechową przejawiajacą się przyspieszonym płytkim oddechem z udziałem skrzydełek nosa i pomocniczych mięśni oddechowych, tachykardią, szarym zabarwieniem powłok i niepokojem. w zaawansowanej postaci ALS z zespołem opuszkowym i rzekomoopuszkowym bardzo precyzyjne badania ruchów gałek ocznych wykazują ich zaburzenia.")
50
Stwardnienie zanikowe boczne. Zanik mięśni obręczy barkowej

51
Dyzartria definicja D y z a r t r i a (łac. dysarthria; w literaturze medycznej: dyzartria), terminem tym określa się: zaburzenia mowy wynikające z uszkodzenia ośrodków i dróg unerwiających narządy mowy. Wskutek uszkodzeń - o różnym stopniu i rozległości powstają zakłócenia w napięciu mięśni biorących udział w akcie mowy, co powoduje zaburzenia kontroli i koordynacji czynności tychże mięśni. Pod pojęciem dyzartrii należy rozumieć szereg objawów, które w zależności od poziomu uszkodzenia sklasyfikowano w odrębne syndromy (Styczek, 1980); zespół zaburzeń oddechowo-fonacyjno-artykulacyjnych, spowodowanych uszkodzeniem ośrodków i dróg unerwiających aparat mówienia (Mitrinowicz-Modrzejewska, 1971). Choć terminem tym często określa się jedynie zaburzenia artykulacji, należy pamiętać, że dochodzi do nich również w wyniku zaburzeń fonacji (generowania dźwięków w obrębie krtani) oraz zaburzeń rezonacji (zmian brzmienia dźwięku zachodzących w nasadzie, tj. w jamie ustnej, nosowej i części jamy gardłowej).
; zespół zaburzeń oddechowo-fonacyjno-artykulacyjnych, spowodowanych uszkodzeniem ośrodków i dróg unerwiających aparat mówienia (Mitrinowicz-Modrzejewska, 1971). Choć terminem tym często określa się jedynie zaburzenia artykulacji, należy pamiętać, że dochodzi do nich również w wyniku zaburzeń fonacji (generowania dźwięków w obrębie krtani) oraz zaburzeń rezonacji (zmian brzmienia dźwięku zachodzących w nasadzie, tj. w jamie ustnej, nosowej i części jamy gardłowej).")
52
Objawy Najważniejszym objawem językowym dyzartrii są zaburzenia artykulacji, co powoduje, że jest ona podobna do dyslalii (podkreślają to niektórzy autorzy, jak np. Leon Kaczmarek, ujmujący dyzartrię w swojej klasyfikacji objawowej dyslalii), jednakże w zakresie przyczyn i innych współwystępujących z dyzartrią objawów (takich jak: zaburzenia fonacji, oddechu, zaburzenia napięcia mięśniowego, współruchy, zaburzenia płynności mowy itd.) różnice są na tyle znaczne, że nawet dla potrzeb kategoryzacji objawowych nie należy łącznie traktować tych zaburzeń. Zaburzenia artykulacji mogą mieć różny stopień nasilenia - od zupełnej niemożności tworzenia artykułowanych dźwięków (anartria) do częściowego upośledzenia tej czynności (dyzartria).
, jednakże w zakresie przyczyn i innych współwystępujących z dyzartrią objawów (takich jak: zaburzenia fonacji, oddechu, zaburzenia napięcia mięśniowego, współruchy, zaburzenia płynności mowy itd.) różnice są na tyle znaczne, że nawet dla potrzeb kategoryzacji objawowych nie należy łącznie traktować tych zaburzeń. Zaburzenia artykulacji mogą mieć różny stopień nasilenia - od zupełnej niemożności tworzenia artykułowanych dźwięków (anartria) do częściowego upośledzenia tej czynności (dyzartria).")
53
Objawy poszczególnych rodzajów dyzartrii w zależności od poziomu uszkodzenia ośrodkowego układu nerwowego Dyzartria korowa Charakteryzuje się zwiększonym napięciem mięśniowym. Występują zaburzenia artykulacji, fonacji i oddechu oraz zmiany w tempie mowy, w melodii i akcentowaniu. Pola ruchowe odpowiedzialne za pracę języka, żuchwy, gardła i krtani znajdują się w dolnej części zakrętu przedśrodkowego w obu półkulach. Obustronne uszkodzenie tych pól powoduje porażenie odpowiedniego narządu mowy, jednostronne - nieznaczne obniżenie jego ruchliwości. Zaburzeniom ulegają bardziej złożone struktury wypowiedzi, co oznacza, że chory nie ma większych trudności z wypowiedzeniem krótkich i prostych wyrazów; trudności występują dopiero podczas wypowiadania wyrazów bardziej skomplikowanych. Dyzartria piramidowa (rzekomoopuszkowa) Występuje zwiększone napięcie mięśni aparatu mowy o charakterze spastycznym, kurczowym (scyzorykowym), które maleje przy powtarzaniu ruchów. Porażenie spastyczne sprawia, że ruchy artykulacyjne są przesadne i nieskoordynowane. Mówienie jest wolne i niepłynne, a wymowa wielu głosek zniekształcona. Widoczne są niedowłady mięśni (m.in. również narządów mowy) oraz współruchy. Zaburzenia mowy zależą od tego, w jakim stopniu i jakie grupy mięśni zostały porażone. Występują: zmiany w tempie i melodii mówienia, zaburzenia oddechu, trudności w wytwarzaniu głosu i w artykulacji, co obserwuje się również w dyzartrii korowej i opuszkowej.
 Występuje zwiększone napięcie mięśni aparatu mowy o charakterze spastycznym, kurczowym (scyzorykowym), które maleje przy powtarzaniu ruchów. Porażenie spastyczne sprawia, że ruchy artykulacyjne są przesadne i nieskoordynowane. Mówienie jest wolne i niepłynne, a wymowa wielu głosek zniekształcona. Widoczne są niedowłady mięśni (m.in. również narządów mowy) oraz współruchy. Zaburzenia mowy zależą od tego, w jakim stopniu i jakie grupy mięśni zostały porażone. Występują: zmiany w tempie i melodii mówienia, zaburzenia oddechu, trudności w wytwarzaniu głosu i w artykulacji, co obserwuje się również w dyzartrii korowej i opuszkowej.")
54
Objawy poszczególnych rodzajów dyzartrii w zależności od poziomu uszkodzenia ośrodkowego układu nerwowego Dyzartria opuszkowa Charakteryzuje się wzmożonym napięciem mięśniowym. Często w akcie mowy obserwuje się współruchy, co sprawia, że ruchy artykulacyjne są przesadne, nieskoordynowane, nieuporządkowane. Porażenie mięśni aparatu mowy może być całkowite lub częściowe. Przy porażeniu częściowym największe zniekształcenia występują w zakresie realizacji głosek wymagających dokładnej koordynacji i zwiększonego napięcia mięśniowego. Występują trudności w żuciu i połykaniu. Czasem pojawia się atrofia mięśni (zazwyczaj języka i mięśnia okrężnego ust). Występuje również drżenie języka. Zaburzenia mowy polegają na (por. Sovak, Styczek, Tarkowski): nieprawidłowym artykułowaniu głosek, zmianach w tempie mowy, melodii i akcentowaniu, zaburzeniach oddechu i fonacji. Najważniejszym objawem zaburzeń dyzartrycznych są zaburzenia artykulacji, przy czym samogłoski są lepiej realizowane niż spółgłoski. W zależności od tego czy przeważa niedowład warg, czy podniebienia, na plan pierwszy wysuwają się trudności w tworzeniu albo spółgłosek wargowych (b, p, w, f), albo podniebiennych (g, k, ch). Natomiast niedowład języka powoduje zaburzenia wymowy głosek d, t, r, s. Częste są substytucje, elizje i deformacje głosek. Mowa dyzartryczna jest więc: niewyraźna, powolna, zamazana, cicha (afoniczna), nosowa. W miarę nasilania się wymienionych objawów mowa staje się coraz bardziej niezrozumiała, bełkotliwa, aż do całkowitej niemożności wydawania artykułowanych dźwięków. Objawy zaburzeń mowy zależą więc od tego, które mięśnie i w jakim stopniu zostały porażone, co powoduje występowanie - poza zaburzeniami artykulacji - również zaburzeń w realizacji płaszczyzny suprasegmentalnej wypowiedzi (rytmu, tempa, melodii, akcentowania), a także trudności w wytwarzaniu głosu (fonacyjne) oraz trudności oddechowe, wynikające z braku regularności oddechu.
. Występuje również drżenie języka. Zaburzenia mowy polegają na (por. Sovak, Styczek, Tarkowski): nieprawidłowym artykułowaniu głosek, zmianach w tempie mowy, melodii i akcentowaniu, zaburzeniach oddechu i fonacji. Najważniejszym objawem zaburzeń dyzartrycznych są zaburzenia artykulacji, przy czym samogłoski są lepiej realizowane niż spółgłoski. W zależności od tego czy przeważa niedowład warg, czy podniebienia, na plan pierwszy wysuwają się trudności w tworzeniu albo spółgłosek wargowych (b, p, w, f), albo podniebiennych (g, k, ch). Natomiast niedowład języka powoduje zaburzenia wymowy głosek d, t, r, s. Częste są substytucje, elizje i deformacje głosek. Mowa dyzartryczna jest więc: niewyraźna, powolna, zamazana, cicha (afoniczna), nosowa. W miarę nasilania się wymienionych objawów mowa staje się coraz bardziej niezrozumiała, bełkotliwa, aż do całkowitej niemożności wydawania artykułowanych dźwięków. Objawy zaburzeń mowy zależą więc od tego, które mięśnie i w jakim stopniu zostały porażone, co powoduje występowanie - poza zaburzeniami artykulacji - również zaburzeń w realizacji płaszczyzny suprasegmentalnej wypowiedzi (rytmu, tempa, melodii, akcentowania), a także trudności w wytwarzaniu głosu (fonacyjne) oraz trudności oddechowe, wynikające z braku regularności oddechu.")
55
Diagnostyka Podstawowy profil biochemiczny i analityczny (płyn mózgowo-rdzeniowy jest w wiekszości pzypadków prawidłowy, podwyższony poziom białka 60-70mg% i cytoza 3-4/1mm3 ) Morfologia krwi z rozmazem i OB. Próby wątrobowe Proteinogram(różnicowanie z neuropatia) Adczyny kiłowe(różnicowanie z amiotrofia w przebiegu kiły) Harmony tarczycy(miopatia, neuropatia czy mielopatia w przebiegu nadczynności tarczycy może przypominać chorobe neuronu ruchowego – niezwykle rzadkie) Aktywność heksaminidazy w leukocytach u ludzi młodych RTG klatki piersiowej i kregosłupa Patologiczne zmiany w EEG EMG – nawet w początkowym okresie choroby może wykazać odnrwieniez obecnością drżeń pęczkowych w kończynie, w której stwierdza się niewielki zanik mięśniowy, zmiany w pozornie niezmienionej kończynie sugerują rozegły obszar chorobowy EMG – daje możliwość oceny rozległości procesu jak i jego dynamiki, umozliwiaja różnicowanie z neuropatiami Mielografia Biopsja mieśnia – cechy odnerwienia, dynamika precesu, wiecej niż jeden mięsien lub mięśnie opuszkowe TK i MRI pozwalaja przede wszystkim na wykluczenie innych niż ALS procesów chorobowych Drobne hipertensyjne zmiany w T2 w tylnej ododze torebki wewnetrznej są wegług niektórych autorów parognomiczne dla ALS W MRI można się spodziewać zmian w odcinku szyjnym rdzenia kregowego próg piramidowych, ponieważ zmainy sa tam najbardziej nasilone Badania spirometryczne oraz gazometria, ułatwiaja bowiem ocenę stopnia niewydolności oddechowej i stanowią o konieczności intubacji Inne badania są mniej specyjiczne(można nadmienić tu immumoenzymatyczne serologiczne, genetyczne), dla tych badan przesłanki istnieją, ale opierają się na słuszności hipotez, którezresztą nie zostały potwierdzone
 Morfologia krwi z rozmazem i OB. Próby wątrobowe. Proteinogram(różnicowanie z neuropatia) Adczyny kiłowe(różnicowanie z amiotrofia w przebiegu kiły) Harmony tarczycy(miopatia, neuropatia czy mielopatia w przebiegu nadczynności tarczycy może przypominać chorobe neuronu ruchowego – niezwykle rzadkie) Aktywność heksaminidazy w leukocytach u ludzi młodych. RTG klatki piersiowej i kregosłupa. Patologiczne zmiany w EEG. EMG – nawet w początkowym okresie choroby może wykazać odnrwieniez obecnością drżeń pęczkowych w kończynie, w której stwierdza się niewielki zanik mięśniowy, zmiany w pozornie niezmienionej kończynie sugerują rozegły obszar chorobowy. EMG – daje możliwość oceny rozległości procesu jak i jego dynamiki, umozliwiaja różnicowanie z neuropatiami. Mielografia. Biopsja mieśnia – cechy odnerwienia, dynamika precesu, wiecej niż jeden mięsien lub mięśnie opuszkowe. TK i MRI pozwalaja przede wszystkim na wykluczenie innych niż ALS procesów chorobowych. Drobne hipertensyjne zmiany w T2 w tylnej ododze torebki wewnetrznej są wegług niektórych autorów parognomiczne dla ALS. W MRI można się spodziewać zmian w odcinku szyjnym rdzenia kregowego próg piramidowych, ponieważ zmainy sa tam najbardziej nasilone. Badania spirometryczne oraz gazometria, ułatwiaja bowiem ocenę stopnia niewydolności oddechowej i stanowią o konieczności intubacji. Inne badania są mniej specyjiczne(można nadmienić tu immumoenzymatyczne serologiczne, genetyczne), dla tych badan przesłanki istnieją, ale opierają się na słuszności hipotez, którezresztą nie zostały potwierdzone.")
56
BADANIE RUCHÓW CZYNNYCH :
polecamy choremu wykonywać ruchy czynnne jednocześnie prawą i lewą kończyną w poszczególnych stawach zwracamy uwagę na zakres i szybkość ruchów ruchy po stronie dominującej są zazwyczaj silniejsze i sprawniejsze badamy kolejno : ruch barku (ku górze, przodowi, tyłowi) ruchy w stawie barkowym (ruchy wyprostowanej k.g w płaszczyźnieczołowej i strzałkowej) ruchy w stawie łokciowym ruchy w stawie nadgarstkowym ruchy palców (zginanie, postowanie, odwodzenie, przywodzenie)
 ruchy w stawie barkowym (ruchy wyprostowanej k.g w płaszczyźnieczołowej i strzałkowej) ruchy w stawie łokciowym. ruchy w stawie nadgarstkowym. ruchy palców (zginanie, postowanie, odwodzenie, przywodzenie)")
57
BADANIE RUCHÓW BIERNYCH I NAPIĘCIA MIĘŚNI :
pacjent powinien mieć odwróconą uwagę np. rozmową podczas badania polecamy choremu rozluźnić mięśnie wykonujemy kolejno ruchy we wszystkich stawach obu kończyn zwracamy uwagę na zakres ruchów i napięcie mięśniowe opór który wyczuwamy podczas wykonywania ruchów biernych wskutek napinania się mięśni jest miarą napięcia mieśniowego ocenę napięcia umożliwia też obmacywanie mięśni w spokoju i podczas ruchów biernych zakres ruchów biernych : ograniczenie zakresu : zmiany w stawach przykurcze (contractura) – rozwijają się w następstwie silnego i długotrwałego napięcia mięśni, lub w następstwie niedowładu napięcie mięśniowe : obniżone : oporu podczas wykonywania ruchów biernych mięśnie zwiotczałe, kontury zatarte, nadmierna ruchomość stawów (możliwe wyprostowanie, lub zgięcie w zakresie przekraczającym granice fizjologiczne) podczas wykonywania szybkich ruchów biernych (potrząsanie) przedramieniem w różnych kierunkach łatwo można zauważyć nadmierne wychylenie ręki stan spastyczny (kurczowy) : spasticitas wzmożenie napięcia mięśniowego typu scyzorykowego podczas wykonywania biernych ruchów szybkich na początku ruchu opór jest największy, po czym dość nagle się i w miarę wykonywania ruchów czasem zupełnie ustępuje towarzyszy uszkodzeniu drogi korowo – rdzeniowej
 – rozwijają się w następstwie silnego i długotrwałego napięcia mięśni, lub w następstwie niedowładu. napięcie mięśniowe : obniżone : oporu podczas wykonywania ruchów biernych. mięśnie zwiotczałe, kontury zatarte, nadmierna ruchomość stawów (możliwe wyprostowanie, lub zgięcie w zakresie przekraczającym granice fizjologiczne) podczas wykonywania szybkich ruchów biernych (potrząsanie) przedramieniem w różnych kierunkach łatwo można zauważyć nadmierne wychylenie ręki. stan spastyczny (kurczowy) : spasticitas. wzmożenie napięcia mięśniowego typu scyzorykowego. podczas wykonywania biernych ruchów szybkich na początku. ruchu opór jest największy, po czym dość nagle się i w miarę. wykonywania ruchów czasem zupełnie ustępuje. towarzyszy uszkodzeniu drogi korowo – rdzeniowej.")
58
BADANIE SIŁY MIĘŚNIOWEJ :
poleca się choremu wykonać ruch w jednym ze stawów, a jednocześnie przeciwstawiamy temu ruchowi opór własnej ręki badamy siłę we wszystkich stawach w kolejności tj. przy badaniu ruchów czynnych osłabienie siły mięśniowej = niedowład (paresis) całkowita niemożność wykonania ruchu = porażenie (paralysis) niedowład w skali bydgoskiej : kończyna górna : 0p. – brak ruchu, lub ślad ruchu 1p. – możliwość wykonania niepełnego zakresu ruchu w odciążeniu 2p. – pełen zakres ruchów bez obciążenia 3p. – pełen zakres ruchów z obciążeniem ręka : 0p. – brak ruchu 1p. – ręka chwytna 2p. – ręka manipulacyjna z pełną opozycją kciuka 3p. – ręka gestowa
 całkowita niemożność wykonania ruchu = porażenie (paralysis) niedowład w skali bydgoskiej : kończyna górna : 0p. – brak ruchu, lub ślad ruchu. 1p. – możliwość wykonania niepełnego zakresu ruchu w odciążeniu. 2p. – pełen zakres ruchów bez obciążenia. 3p. – pełen zakres ruchów z obciążeniem. ręka : 0p. – brak ruchu. 1p. – ręka chwytna. 2p. – ręka manipulacyjna z pełną opozycją kciuka. 3p. – ręka gestowa.")
59
wzmacnianie odruchów :
BADANIE ODRUCHÓW : wzmacnianie odruchów : np. pacjent zaciska zęby odruch z m. brachioradialis : ośrodek C5 – C6 (n. radialis) ramię chorego jest przywiedzione, przedramię zgięte w stos. do ramienia pod kątem ok. 120o , ręka w ułożeniu pośrednim między supinacją i pronacją lekarz ujmuje silnie lewą ręką brzeg łokciowy ręki badanego i uderza młotkiem w wyrostek rylcowaty k. promieniowej następuje zgięcie w stawie łokciowym (skurcz m. brachioradialis, m. biceps brachii i m. brachialis internus) po uderzeniu w wyrostek rylcowaty często równocześnie stwierdza się odruch Jacobsona : polega na zgięciu palców obustronny jest fizjologiczny, a jednostronny patologiczny odruch z m. biceps brachii : ośrodek C5 – C6 (n. musculocutaneus) ułożenie kończyny jak w odruchu z m. brachioradialis, ale ręka jest w całkowitej supinacji i opiera się na dłoni lekarza wyczuwamy ścięgno m. dwugłowego i uderzamy w nie młotkiem powoduje to skurcz m. dwugłowego i zgięcie w stawie łokciowym odruch z m. triceps brachii : ośrodek C7 (n. radialis) unosimy ramię ku górze do kąta 70 – 75o w stosunku do tułowia zginamy kończynę w stawie łokciowym do kąta nieco > 90o podtrzymujemy ramię w ten sposób, aby zwisało ku dołowi uderzamy w ścięgno m. trójgłowego tuż powyżej wyrostka łokciowego (olecranon) występuje skurcz m. trójgłowego i ruch wyprostny przedramienia inny sposób : ramię unosimy do poziomu i podtrzymujemy tak aby zwisające swobodnie przedramię tworzyło z nim kąt prosty, dalej tjw.
 ramię chorego jest przywiedzione, przedramię zgięte w stos. do ramienia. pod kątem ok. 120o , ręka w ułożeniu pośrednim między supinacją i pronacją. lekarz ujmuje silnie lewą ręką brzeg łokciowy ręki badanego i uderza młotkiem w wyrostek rylcowaty k. promieniowej. następuje zgięcie w stawie łokciowym (skurcz m. brachioradialis, m. biceps brachii i m. brachialis internus) po uderzeniu w wyrostek rylcowaty często równocześnie stwierdza się. odruch Jacobsona : polega na zgięciu palców. obustronny jest fizjologiczny, a jednostronny patologiczny. odruch z m. biceps brachii : ośrodek C5 – C6 (n. musculocutaneus) ułożenie kończyny jak w odruchu z m. brachioradialis, ale ręka jest. w całkowitej supinacji i opiera się na dłoni lekarza. wyczuwamy ścięgno m. dwugłowego i uderzamy w nie młotkiem. powoduje to skurcz m. dwugłowego i zgięcie w stawie łokciowym. odruch z m. triceps brachii : ośrodek C7 (n. radialis) unosimy ramię ku górze do kąta 70 – 75o w stosunku do tułowia. zginamy kończynę w stawie łokciowym do kąta nieco > 90o. podtrzymujemy ramię w ten sposób, aby zwisało ku dołowi. uderzamy w ścięgno m. trójgłowego tuż powyżej wyrostka łokciowego. (olecranon) występuje skurcz m. trójgłowego i ruch wyprostny przedramienia. inny sposób : ramię unosimy do poziomu i podtrzymujemy tak aby. zwisające swobodnie przedramię tworzyło z nim kąt prosty, dalej tjw.")
60
odruch z grupy odruchów podstawowych
odruch Meyera : odruch z grupy odruchów podstawowych przywiedzenie i wyprostowanie kciuka podczas silnego zgięcia w stawie podstawowym III, lub IV palca ręki obusronny brak może być fizjologiczny jednostronny brak : uszkodzenie drogi korowo – rdzeniowej odruch chwytny : występuje jednostronnie w przypadku guza płata czołowego przeciwstronnie do uszkodzenia chory mimowolnie chwyta przedmiot, który mu się wsadzi do ręki usunięcie przedmiotu napotyka trudności chory sam nie może czynnie wypuścić przedmiotu zmiany patologiczne : żywe i wygórowane odruchy : w zespole piramidowym osłabienie, lub zniesienie odruchów : ogólne : neuropatia obwodowa zespół móżdżkowy odosobnione : uszkodzenie obwodowego neuronu ruchowego anizorefleksja = asymetria odruchów odruch odwrócony (paradoksalny) : odruch w danym miejscu jest zniesiony, lecz zarazem przeniesiony na niższy poziom poziom zniesionego odruchu odpowiada miejscu zmian chorobowych np. odruch z m. dwugłowego zniesiony, lecz uderzenie wywołuje skurcz m. trójgłowego jest objawem uszkodzenia obwodowego neuronu ruchowego na danym poziomie np.C5 połączonego z uszkodzeniem ośrodkowego neuronu ruchowego poniżej tego poziomu (zajęcie rdzenia kręgowego na poziomie zniesionego odruchu) powolne rozlużnianie mięśni po wywołaniu odruchu : objaw niedoczynności tarczycy
 : odruch w danym miejscu jest zniesiony, lecz zarazem przeniesiony na niższy poziom. poziom zniesionego odruchu odpowiada miejscu zmian chorobowych. np. odruch z m. dwugłowego zniesiony, lecz uderzenie wywołuje skurcz m. trójgłowego. jest objawem uszkodzenia obwodowego neuronu ruchowego na. danym poziomie np.C5 połączonego z uszkodzeniem ośrodkowego. neuronu ruchowego poniżej tego poziomu (zajęcie rdzenia kręgowego. na poziomie zniesionego odruchu) powolne rozlużnianie mięśni po wywołaniu odruchu : objaw niedoczynności tarczycy.")
61
BADANIE SIŁY MIĘŚNIOWEJ KOŃCZYN DOLNYCH:
tj. kończyna górna niedowład w skali bydgoskiej : 0p. – brak ruchu, lub ślad ruchu 1p. – niepełny zakrres ruchów w odciążeniu 2p. – pełny zakres ruchów bez obciążenia 3p. – pełny zakres ruchów z obciążeniem próba Barrego : choremu leżącemu na brzuchu zginamy obie kończyny dolne w stawach kolanowych pod kątem prostym i polecamy utrzymać kończyny w tej pozycji po stronie niedowładu kończyna opada (można wykryć lekki niedowład) inne sposoby wykrycia lekkiego niedowładu : leżący chory kładzie skrzyżowane ręce na klatce piersiowej, rozsuwa nieco kończyny dolne i siada ludzie zdrowi opierają się piętami o posłanie jeśli jest niedowład kończyna się unosi uniesienie 1, lub obu kończyn może wystąpić w chorobach móżdżku
 inne sposoby wykrycia lekkiego niedowładu : leżący chory kładzie skrzyżowane ręce na klatce piersiowej, rozsuwa nieco kończyny dolne i siada. ludzie zdrowi opierają się piętami o posłanie. jeśli jest niedowład kończyna się unosi uniesienie 1, lub obu kończyn może wystąpić w chorobach móżdżku.")
62
odruch kolanowy (rzepkowy) : - ośrodek : L2 – L4 (n. udowy)
BADANIE ODRUCHÓW : odruchy można wzmocnić przez zaciśnięcie pięści, lub złączenie dłoni i ich rozciąganie, liczenie patrzenie w sufit itp. brak odruchu można stwierdzić dopiero po wielokrotnym badaniu ze wzmocnieniem odruch kolanowy (rzepkowy) : - ośrodek : L2 – L4 (n. udowy) chory siedzi na krześle ze stopami opartymi o podłogę, kończyny zgięte w stawach kolanowych pod kątem 100o lekarz kładzie rękę na udzie chorego i uderza młotkiem tuż poniżej rzepki w ścięgno m. qadriceps widoczny i wyczuwalny jest skurcz m. qadriceps oraz ruch wyprostny w stawie kolanowym niekiedy występuje równoczesne przywiedzenie kończyny badanej, czasem obu kończyn (skurcz przywodzicieli) można też wywołać gdy nogi swobodnie zwisają u chorych leżących kończyny zginamy w stawach kolanowych pod kątem 120 – 130o, stopy opierają się całą powierzchnią o posłanie, chory rozlużnia mięśnie, lekarz podtrzymuje obie kończyny jedną ręką, którą umieszcza pod kolanami badanego odruch skokowy : ośrodek S1 – S2 (n. kulszowy) chory klęczy na krześle, lub łóżku tak, aby tylko stopa luźno zwisała poza brzeg krzesła, lub łóżka uderzamy młotkiem w ścięgno Achillesa następuje skurcz m. triceps surae i zgięcie podeszwowe stopy u chorych leżących odruch możemy wywołać po odwiedzeniu kończyny i zgięciu w stawie kolanowym w ten sposób, aby podudzie tej kończyny krzyżowało się z podudziem drugiej kończyny, która pozostaje wyprostowana odruch podeszwowy : drażnienie skóry podeszwy wywołuje zgięcie podeszwowe palucha należy drażnić skórę zewnętrznej powierzchni podeszwy wdłuż linii biegnącej od pięty ku okolicy nasady V palca zgięciu palucha towarzyszy zgięcie pozostałych palców stopy reakcja obronna : paluch prostuje się i pozostałe palce też, a stopa zgina się w kostce należy powtórzyć badanie delikatniej zniesienie odruchu (areflexia plantis) : paluch ani się nie prostje, ani nie zgina może wystąpić w wyniku oziębienia stopy
 : - ośrodek : L2 – L4 (n. udowy) chory siedzi na krześle ze stopami opartymi o podłogę, kończyny zgięte w stawach kolanowych pod kątem 100o. lekarz kładzie rękę na udzie chorego i uderza młotkiem tuż poniżej rzepki w ścięgno m. qadriceps. widoczny i wyczuwalny jest skurcz m. qadriceps oraz ruch wyprostny w stawie kolanowym. niekiedy występuje równoczesne przywiedzenie kończyny badanej, czasem obu kończyn (skurcz przywodzicieli) można też wywołać gdy nogi swobodnie zwisają. u chorych leżących kończyny zginamy w stawach kolanowych pod kątem 120 – 130o, stopy opierają się całą powierzchnią o posłanie, chory rozlużnia mięśnie, lekarz podtrzymuje obie kończyny jedną ręką, którą umieszcza pod kolanami badanego. odruch skokowy : ośrodek S1 – S2 (n. kulszowy) chory klęczy na krześle, lub łóżku tak, aby tylko stopa luźno zwisała poza brzeg krzesła, lub łóżka. uderzamy młotkiem w ścięgno Achillesa. następuje skurcz m. triceps surae i zgięcie podeszwowe stopy. u chorych leżących odruch możemy wywołać po odwiedzeniu kończyny i zgięciu w stawie kolanowym w ten sposób, aby podudzie tej kończyny krzyżowało się z podudziem drugiej kończyny, która pozostaje wyprostowana. odruch podeszwowy : drażnienie skóry podeszwy wywołuje zgięcie podeszwowe palucha. należy drażnić skórę zewnętrznej powierzchni podeszwy wdłuż linii biegnącej od pięty ku okolicy nasady V palca. zgięciu palucha towarzyszy zgięcie pozostałych palców stopy. reakcja obronna : paluch prostuje się i pozostałe palce też, a stopa zgina się w kostce. należy powtórzyć badanie delikatniej. zniesienie odruchu (areflexia plantis) : paluch ani się nie prostje, ani nie zgina. może wystąpić w wyniku oziębienia stopy.")
63
wywołujemy tj. podeszwowy występuje grzbietowe zgięcie palucha
odruch Babińskiego : odruch patologiczny wywołujemy tj. podeszwowy występuje grzbietowe zgięcie palucha (+) objaw Babińskiego : uszkodzenie drogi korowo – rdzeniowej w 1 – 2 rż. fizjologiczny łącznie z odruchem Babińskiego często występuje wachlarzowate odwiedzenie palców stopy (objaw wachlarza) może wystąpić zgięcie grzbietowe stopy, zgięcie w stawie kolanowymi biodrowym wyraz automatyzmu rdzeniowego odruch Rossolimo : uderzamy szybko i dość energicznie opuszkami palców, lub młotkiem w opuszki palców stopy rozciąginięcie zginaczy palców stopy i zgięcie podeszwowe palców stopy (+) odruch Rossolimo : jeden z pierwszych objawów SM niekiedy u zdrowych klonusy : gdy odruchy są bardzo wzmożone towarzyszą im klonusy o klonusie mówimy gdy wystąpią co najmniej 3 rytmiczne skurcze klonus rzepki (rzepkotrząs) : można go uzyskać nagłym ruchem biernym rzepki ku dołowi silnie ująć rzepkę miedzy kciuk i palec wskazujący i pociągnąć ku dołowi w m. quadriceps występują rytmiczne skurcze co powoduje ruchy rzepki ku górze i ku dołowi klonus stopy (stopotrząs) : po nagłym wykonaniu grzbietowego zgięcia stopy kończyna zgięta w stawie kolanowym i stopa podtrzymywana w zgięciu ręką badającego niekiedy klonus pojawia się po wywołaniu odruchu skokowego
 objaw Babińskiego : uszkodzenie drogi korowo – rdzeniowej. w 1 – 2 rż. fizjologiczny. łącznie z odruchem Babińskiego często występuje wachlarzowate odwiedzenie palców stopy (objaw wachlarza) może wystąpić zgięcie grzbietowe stopy, zgięcie w stawie kolanowymi biodrowym. wyraz automatyzmu rdzeniowego. odruch Rossolimo : uderzamy szybko i dość energicznie opuszkami palców, lub młotkiem w opuszki palców stopy. rozciąginięcie zginaczy palców stopy i zgięcie podeszwowe palców stopy. (+) odruch Rossolimo : jeden z pierwszych objawów SM. niekiedy u zdrowych. klonusy : gdy odruchy są bardzo wzmożone towarzyszą im klonusy. o klonusie mówimy gdy wystąpią co najmniej 3 rytmiczne skurcze. klonus rzepki (rzepkotrząs) : można go uzyskać nagłym ruchem biernym rzepki ku dołowi. silnie ująć rzepkę miedzy kciuk i palec wskazujący i pociągnąć ku dołowi. w m. quadriceps występują rytmiczne skurcze co powoduje ruchy rzepki ku górze i ku dołowi. klonus stopy (stopotrząs) : po nagłym wykonaniu grzbietowego zgięcia stopy. kończyna zgięta w stawie kolanowym i stopa podtrzymywana w zgięciu ręką badającego. niekiedy klonus pojawia się po wywołaniu odruchu skokowego.")
64
Zarys różnicowania

66
Różnicowanie choroby naczyniowe, guzy mózgu i pnia mózgu,
chorobę Parkinsona, choroby móżdżku, neuropatie, choroby neuronów ruchowych, miopatie, zapalenia mięśni, miastenię i inne choroby układu ruchu

67
Rokowanie Stwardnienie zanikowe boczne (ALS) zalicza się do chorób, wobec których neurolog jest całkowicie bezradny. Pesymizm poteguje prognoze : choroba jest nieuchronnie postepująca, prowadząc w ciągu kilku lat do zgonu, poprzedzonego okresem pełnej zależności od osób trzecich i specjalistycznej aparatury do sztucznego oddechu. Okres przeżycia od momentu wykrecia choroby uzależniony jest, jak się wydaje(nap przykładzie Hawkinga) niemal zupełnie od cech indywidualnych chorego tj. od konstytucji genetycznej. Czas wykrycia choroby w żaden sposób nie wpływa na zwiekszenie przeżywalnności. Ogromną rolę w opiece nad chorym odgrywa leczenie pbjawowe , umiarkowana rehabilitacja i staranna higiena chorego(drzewa oskrzelowego). Chory umiera z powodów internistycznych , nie neurologicznych. Istnieje mozliwosc długoterminowego utrzymywania chorego przy zyciu, jednak wymaga to współdziałania zespołu wielospecjalistycznego i wygospodarowania adekwatnych(nie małych) srodków finansowych. Wobec powyższego można stwierdzic, że rokowanie jest skrajnie niepomyslne.
 zalicza się do chorób, wobec których neurolog jest całkowicie bezradny. Pesymizm poteguje prognoze : choroba jest nieuchronnie postepująca, prowadząc w ciągu kilku lat do zgonu, poprzedzonego okresem pełnej zależności od osób trzecich i specjalistycznej aparatury do sztucznego oddechu. Okres przeżycia od momentu wykrecia choroby uzależniony jest, jak się wydaje(nap przykładzie Hawkinga) niemal zupełnie od cech indywidualnych chorego tj. od konstytucji genetycznej. Czas wykrycia choroby w żaden sposób nie wpływa na zwiekszenie przeżywalnności. Ogromną rolę w opiece nad chorym odgrywa leczenie pbjawowe , umiarkowana rehabilitacja i staranna higiena chorego(drzewa oskrzelowego). Chory umiera z powodów internistycznych , nie neurologicznych. Istnieje mozliwosc długoterminowego utrzymywania chorego przy zyciu, jednak wymaga to współdziałania zespołu wielospecjalistycznego i wygospodarowania adekwatnych(nie małych) srodków finansowych. Wobec powyższego można stwierdzic, że rokowanie jest skrajnie niepomyslne.")
68
Leczenie Farmakologiczne (rys historyczny ?): amantadyna, izoprenozyna, penicylamina, steroidy, interferon, wit. E skojarzona z deprenilem(DEPRENYL: MAO-B INHIBITOR EXTRAORDINAIRE. ... DEPRENYL: CATECHOLAMINE ACTIVITY ENHANCER), lub skojarzony z wyciągami z trzustki, wit. B12, baklofen i inne , różne metody leczenia wynikały z nowej, proponowanej w danym okresie, hipotezy dotyczącej etiopatogenezy choroby, pomimo faktu znacznej liczebności okazały się nieskuteczne Czynniki wzrostu : prowadzono próby nad rHCNF ( recombinant human ciliary neurotrophic factor - cytokina), pomimo oiecujących efektów uzyskanych na materiale doświadczalnym (z przecięciem aksonu, uszkodzeniem komórek, hodowla tkankową) nie udała się próba na ludziach, co wiecej wystąpiły znaczne efekty uboczne(ALSCNTF treatment Study Group 1996), BDNF, GDNF, GF I i II
: amantadyna, izoprenozyna, penicylamina, steroidy, interferon, wit. E skojarzona z deprenilem(DEPRENYL: MAO-B INHIBITOR EXTRAORDINAIRE. ... DEPRENYL: CATECHOLAMINE ACTIVITY ENHANCER), lub skojarzony z wyciągami z trzustki, wit. B12, baklofen i inne , różne metody leczenia wynikały z nowej, proponowanej w danym okresie, hipotezy dotyczącej etiopatogenezy choroby, pomimo faktu znacznej liczebności okazały się nieskuteczne. Czynniki wzrostu : prowadzono próby nad rHCNF ( recombinant human ciliary neurotrophic factor - cytokina), pomimo oiecujących efektów uzyskanych na materiale doświadczalnym (z przecięciem aksonu, uszkodzeniem komórek, hodowla tkankową) nie udała się próba na ludziach, co wiecej wystąpiły znaczne efekty uboczne(ALSCNTF treatment Study Group 1996), BDNF, GDNF, GF I i II.")
69
Leczenie Riluzole – skuteczność leku polega na przedłużeniu życia o 2-3 miesiace, poza tym nie ma żadnego wpływu! „According to recent reports, early diagnosis and initiation of drug therapy appears to be the best way to prolong functional survival in patients with amyotrophic lateral sclerosis (ALS). Clinical trials with Rilutek, the only drug approved for use in ALS, have produced a slowed disease progression and a 30% increase in survival compared with placebo.” Publication title:Geriatrics. Duluth: Feb 1998. Vol. 53, Iss. 2; pg. 19, 1 pgs Gabapentin – final trial underway Other therapeutic approaches to ALS involve neurotrophic factors. Preclinical studies with both ciliary neurotrophic factor and brain-derived neurotrophic factor (BDNF) were particularly promising, but large multicenter controlled trials of subcutaneous administration produced no beneficial effect upon the disease. Promising studies with intrathecal BDNF and glialderived neurotrophic factor (GDNF) are underway. Finally, encouraging results from the US trial of Insulinlike growth factor (IGF-1), which show that the decline of function and quality of life for patients with ALS is slowing, are currently under review at the FDA. Komórki macierzyste : w fazie badań Objawowe Antycholinergiczne , naswietlanie slinianek w skrajnych przypadkach chinina – w przypadku kurczu mięśni
. Clinical trials with Rilutek, the only drug approved for use in ALS, have produced a slowed disease progression and a 30% increase in survival compared with placebo. Publication title:Geriatrics. Duluth: Feb Vol. 53, Iss. 2; pg. 19, 1 pgs. Gabapentin – final trial underway. Other therapeutic approaches to ALS involve neurotrophic factors. Preclinical studies with both ciliary neurotrophic factor and brain-derived neurotrophic factor (BDNF) were particularly promising, but large multicenter controlled trials of subcutaneous administration produced no beneficial effect upon the disease. Promising studies with intrathecal BDNF and glialderived neurotrophic factor (GDNF) are underway. Finally, encouraging results from the US trial of Insulinlike growth factor (IGF-1), which show that the decline of function and quality of life for patients with ALS is slowing, are currently under review at the FDA. Komórki macierzyste : w fazie badań. Objawowe. Antycholinergiczne , naswietlanie slinianek w skrajnych przypadkach chinina – w przypadku kurczu mięśni.")
70
amantadine antiparkinsonicum, virustaticum N04BB
Działanie: Syntetyczna trójpierścieniowa amina. Ułatwia uwalnianie dopaminy na poziomie prążkowia, zmniejsza objawy jej niedoboru w chorobie Parkinsona - akinezję i w mniejszym stopniu drżenie. Ma również działanie wirusostatyczne - hamuje uwalnianie materiału genetycznego wirusa grypy typu A z nukleokapsydu do komórki i dalsze etapy jego replikacji. Bardzo dobrze się wchłania z przewodu pokarmowego, tmax wynosi 4 h. t1/2 wynosi 11,8 h, u młodych osób, 28,9 h u osób w podeszłym wieku, w przypadku niewydolności nerek 18,5-33,8 h. Klirens nerkowy amantadyny wynosi ok. 389 ml/min. Wydalana jest w stanie niezmienionym z moczem. Coraz częściej izoluje się od chorych szczepy wirusa grypy typu A całkowicie oporne na amantadynę. Podawanie zapobiegawcze i leczenie grypy u dzieci dłużej niż 3-4 dni zwiększa możliwość rozwoju oporności na lek. Wskazania: P.o. Choroba Parkinsona. Zapobieganie zakażeniu wirusem grypy u osób szczególnie narażonych, z przeciwwskazaniami do szczepień. Podana w pierwszym okresie choroby zmniejsza nasilenie objawów grypy. Ponieważ amantadyna nie zmienia odpowiedzi immunologicznej na wirusa typu A, może być stosowana równocześnie ze szczepionką u osób, u których może wystąpić słaba odpowiedź immunologiczna. I.v. w chorobie Parkinsona w stanach zagrożenia życia lub znacznym nasileniu objawów na początku leczenia, nerwobóle w półpaścu, poprawa czuwania i stanu świadomości (np. po urazach). Przeciwwskazania: Bezwzględne: nadwrażliwość na lek; względne: choroba wrzodowa, padaczka, ciężka psychoza, niewydolność serca, hipotonia ortostatyczna, niewydolność nerek i wątroby. Interakcje: Nasila działania niepożądane leków działających antycholinergicznie oraz działanie leków stymulujących OUN. W skojarzeniu z lewodopą mogą wystąpić reakcje psychotyczne. Kotrimoksazol, chinina oraz chinidyna mogą zmniejszać klirens nerkowy amantadyny i nasilać działania niepożądane. Kwaśny odczyn moczu zwiększa wydalanie leku. Triamteren i hydrochlorotiazyd mogą zwiększać stężenia amantadyny w osoczu. Brak interakcji ze szczepionką przeciw wirusowi grypy. Działanie niepożądane: Zawroty głowy, bezsenność, rozdrażnienie. Rzadko depresja, niepokój, omamy, splątanie, zaburzenia ze strony przewodu pokarmowego, bóle głowy, hipotonia ortostatyczna, zaburzenia koncentracji, suchość ust, zaparcia. Ciąża i laktacja: Kategoria C. Lek teratogenny. Nie stosować w okresie karmienia piersią. Dawkowanie: P.o. W chorobie Parkinsona początkowo 100 mg raz na dobę przez 4-7 dni, następnie w razie potrzeby mg/d w dawkach podzielonych, maksymalnie 400 mg/d. W zapobieganiu grypie w razie udokumentowanej epidemii profilaktycznie 100 mg 2 razy na dobę, osobom chorym przez pierwsze 5 dni w dawce 100 mg 2 razy na dobę, po 65. rż. 100 mg raz na dobę. Dzieci do 9. rż. 4-9 mg/kg mc./d, nie więcej niż 150 mg/d; po 9. rż. 100 mg 2 razy na dobę. W niewydolności nerek dawki ustala się indywidualnie w zależności od klirensu kreatyniny. Klirens kreatyniny ml/min/1,73 m2 pc mg 2 razy na dobę, klirens kreatyniny ml/min/1,73 m2 pc. - pierwsza dawka 200 mg, a następnie 100 mg raz na dobę, klirens do 30 ml/min/1,73 m2 pc mg 2 razy w tyg., klirens ml/min/1,73 m2 pc mg 3 razy w tyg., klirens do 15 ml/min/1,73 m2 pc. - dawka na przemian wynosi 100 mg i 200 mg co 7 dni, chorzy dializowani na przemian 200 mg i 100 mg co 7 dni. I.v mg/d w powolnym (55 kropli/min) wlewie, w wyjątkowych przypadkach dawkę można zwiększyć do 600 mg/d; nagłe przerwanie leczenia grozi zaostrzeniem choroby. Uwagi: Lek w postaci doustnej podawać po posiłkach. Nie podawać na noc. Około 1/2 h po podaniu należy chorego sprowokować do kaszlu, ewentualnie odessać zalegającą wydzielinę. Podczas leczenia nie należy prowadzić pojazdów mechanicznych, obsługiwać maszyn, spożywać alkoholu. Preparaty amantadine
. Przeciwwskazania: Bezwzględne: nadwrażliwość na lek; względne: choroba wrzodowa, padaczka, ciężka psychoza, niewydolność serca, hipotonia ortostatyczna, niewydolność nerek i wątroby. Interakcje: Nasila działania niepożądane leków działających antycholinergicznie oraz działanie leków stymulujących OUN. W skojarzeniu z lewodopą mogą wystąpić reakcje psychotyczne. Kotrimoksazol, chinina oraz chinidyna mogą zmniejszać klirens nerkowy amantadyny i nasilać działania niepożądane. Kwaśny odczyn moczu zwiększa wydalanie leku. Triamteren i hydrochlorotiazyd mogą zwiększać stężenia amantadyny w osoczu. Brak interakcji ze szczepionką przeciw wirusowi grypy. Działanie niepożądane: Zawroty głowy, bezsenność, rozdrażnienie. Rzadko depresja, niepokój, omamy, splątanie, zaburzenia ze strony przewodu pokarmowego, bóle głowy, hipotonia ortostatyczna, zaburzenia koncentracji, suchość ust, zaparcia. Ciąża i laktacja: Kategoria C. Lek teratogenny. Nie stosować w okresie karmienia piersią. Dawkowanie: P.o. W chorobie Parkinsona początkowo 100 mg raz na dobę przez 4-7 dni, następnie w razie potrzeby mg/d w dawkach podzielonych, maksymalnie 400 mg/d. W zapobieganiu grypie w razie udokumentowanej epidemii profilaktycznie 100 mg 2 razy na dobę, osobom chorym przez pierwsze 5 dni w dawce 100 mg 2 razy na dobę, po 65. rż. 100 mg raz na dobę. Dzieci do 9. rż. 4-9 mg/kg mc./d, nie więcej niż 150 mg/d; po 9. rż. 100 mg 2 razy na dobę. W niewydolności nerek dawki ustala się indywidualnie w zależności od klirensu kreatyniny. Klirens kreatyniny ml/min/1,73 m2 pc mg 2 razy na dobę, klirens kreatyniny ml/min/1,73 m2 pc. - pierwsza dawka 200 mg, a następnie 100 mg raz na dobę, klirens do 30 ml/min/1,73 m2 pc mg 2 razy w tyg., klirens ml/min/1,73 m2 pc mg 3 razy w tyg., klirens do 15 ml/min/1,73 m2 pc. - dawka na przemian wynosi 100 mg i 200 mg co 7 dni, chorzy dializowani na przemian 200 mg i 100 mg co 7 dni. I.v mg/d w powolnym (55 kropli/min) wlewie, w wyjątkowych przypadkach dawkę można zwiększyć do 600 mg/d; nagłe przerwanie leczenia grozi zaostrzeniem choroby. Uwagi: Lek w postaci doustnej podawać po posiłkach. Nie podawać na noc. Około 1/2 h po podaniu należy chorego sprowokować do kaszlu, ewentualnie odessać zalegającą wydzielinę. Podczas leczenia nie należy prowadzić pojazdów mechanicznych, obsługiwać maszyn, spożywać alkoholu. Preparaty amantadine.")
71
penicillamine antidotum, antirheumaticum M01CC
Działanie: Pochodna penicyliny. Ma właściwości chelatujące - tworzy trwałe, rozpuszczalne w wodzie wiązania kompleksowe z jonami miedzi, ołowiu, rtęci, kadmu, talu, cynku, niklu, złota i żelaza. Powoduje również zmniejszenie wchłaniania jonów miedzi z przewodu pokarmowego, rozbija kompleksy immunoglobulin odkładające się w tkankach w przebiegu reumatoidalnego zapalenia stawów i innych chorób tkanki łącznej, a także spowalnia procesy tworzenia wiązań pomiędzy cząsteczkami kolagenu. Chelatowanie metali wchodzących w skład enzymów - metaloproteinaz - to kolejny możliwy mechanizm działania leku. Penicylamina hamuje aktywność limfocytów T (pozostając bez wpływu na limfocyty B). Lek wchłania się dobrze, osiągając maksymalne stężenie w osoczu 2 h po podaniu p.o. Metabolizm przebiega w dwóch etapach: t1/2 pierwszej fazy wynosi 1 h; drugiej - 5 h; 80% leku wydala się z kałem i moczem w przeciągu 48 h po podaniu, natomiast śladowe ilości mogą pozostawać w osoczu przez kilka do kilkunastu dni. Wskazania: Zapobieganie zwyrodnieniu soczewkowo-wątrobowemu (choroba Wilsona) i jego leczenie. Leczenie zatruć ołowiem, cynkiem, talem, miedzią i złotem, cystynurii oraz reumatoidalnego zapalenia stawów, w którym inne metody leczenia okazały się nieskuteczne (poprawa kliniczna może nastąpić po 1-3 mies. stosowania penicylaminy). Przeciwwskazania: Nadwrażliwość na preparat lub penicylinę, niewydolność nerek, choroby tkanki łącznej z wyjątkiem reumatoidalnego zapalenia stawów, myasthenia gravis. Stosowanie penicylaminy jest przeciwwskazane u chorych, u których wystąpił białkomocz po leczeniu preparatami złota lub niedokrwistość aplastyczna czy agranulocytoza po uprzednim leczeniu penicylaminą. Interakcje: Penicylamina tworzy trwałe związki z wieloma metalami ciężkimi. Działanie niepożądane: Występują często i mogą mieć poważny przebieg, niekiedy prowadzący nawet do zgonu. Do najczęstszych należą reakcje alergiczne: świąd, osutka, pęcherzyca, gorączka, bóle stawów, powiększenie obwodowych węzłów chłonnych, toczeń polekowy, złuszczające zapalenie skóry, pokrzywka, wędrujące zapalenie stawów, zapalenie błony maziowej stawów, astma oskrzelowa; zapalenie gruczołu tarczowego; ze strony przewodu pokarmowego: zmniejszenie łaknienia, zapalenie języka z metalicznym posmakiem w ustach, dziąseł i błony śluzowej jamy ustnej, bóle brzucha, nudności, wymioty, biegunka, uczynnienie choroby wrzodowej żołądka, wewnątrzwątrobowy zastój żółci; objawy te ustępują po odstawieniu leku; hematologiczne: upośledzenie czynności szpiku kostnego, zwiększenie lub zmniejszenie liczby leukocytów i płytek krwi, eozynofilia, agranulocytoza, niedokrwistość aplastyczna, hemolityczna i niedoborowa; nerkowe: białkomocz, krwinkomocz, zespół nerczycowy, zespół Goodpasteure'a; ze strony OUN: zapalenie nerwu wzrokowego, zaburzenia widzenia, szum w uszach, obwodowe neuropatie czuciowe i ruchowe, zaburzenia psychiczne; inne: myasthenia gravis, zakrzepowe zapalenie żył, wypadanie włosów, liszaj płaski, zapalenie wielomięśniowe, przerost gruczołu sutkowego, zapalenie oskrzeli, śródmiąższowe zapalenie płuc. Rozpad czynnika reumatoidalnego spowodowany penicylaminą może wywołać powstanie nowych przeciwciał, powodujących odmiedniczkowe zapalenie nerek, zapalenie mięśni lub zapalenie m. sercowego. Ciąża i laktacja: Nie stosować w ciąży (wyjątek choroba Wilsona - dawkę zmniejszyć do 1 g/d). Nie stosować w okresie karmienia piersią. Dawkowanie: Penicylaminę należy podawać 1 h przed posiłkiem lub 2 h po nim, zachowując przynajmniej godzinny odstęp po podaniu innych leków. Choroba Wilsona. Dawkowanie ustala się w zależności od stanu chorego i dobowej ilości miedzi wydalanej z moczem. Najczęściej rozpoczyna się od dobowej dawki 1 g, którą stopniowo zwiększa się do 2 g/d. Przewlekłe zatrucie ołowiem. Dorośli: 0,9-1,5 g/d w 3 dawkach podzielonych. Dzieci: mg/kg mc./d. Cystynuria. Leczenie rozpoczyna się od 250 mg/d., a następnie powoli zwiększa się dawkę. Dorośli: 2 g/d. Dzieci: mg/kg mc./d w 4 dawkach podzielonych. Reumatoidalne zapalenie stawów przez 1. mies. leczenia stosuje się 125 lub 250 mg/d; następnie dawkę zwiększa się co mies. o mg. Jeśli objawy kliniczne ustępują, leczenie kontynuuje się za pomocą najmniejszej skutecznej dawki. W razie utrzymywania się objawów klinicznych po 3 mies. stosowania dawki mg/d należy zwiększyć ją o kolejne 250 mg co mies. przez kolejne 3 mies. leczenia. Leczenie podtrzymujące: dawkowanie ustala się indywidualnie; najczęściej podaje się mg/d. Jeśli po 4-6 mies. leczenia penicylaminą w dawce max. 0,75 g/d nie występuje poprawa kliniczna, należy zrezygnować ze stosowania leku. Uwagi: Ze względu na częste występowanie działań niepożądanych i toksycznych penicylaminy zaleca się wykonywanie częstych badań kontrolnych morfologii krwi, jonogramu i badania ogólnego moczu - początkowo co 3 dni, następnie raz w tyg. W wypadku wystąpienia działań niepożądanych leczenie należy przerwać. Po ustąpieniu działań niepożądanych leczenie podejmuje się na nowo, rozpoczynając od najmniejszej dawki leku. Podczas leczenia za pomocą penicylaminy wskazane jest jednoczesne podawanie wit. B6. Jeśli preparaty złota okazały się nieskuteczne w leczeniu reumatoidalnego zapalenia stawów, leczenie penicylaminą można rozpocząć nie wcześniej niż 6 mies. po ich odstawieniu.
. Lek wchłania się dobrze, osiągając maksymalne stężenie w osoczu 2 h po podaniu p.o. Metabolizm przebiega w dwóch etapach: t1/2 pierwszej fazy wynosi 1 h; drugiej - 5 h; 80% leku wydala się z kałem i moczem w przeciągu 48 h po podaniu, natomiast śladowe ilości mogą pozostawać w osoczu przez kilka do kilkunastu dni. Wskazania: Zapobieganie zwyrodnieniu soczewkowo-wątrobowemu (choroba Wilsona) i jego leczenie. Leczenie zatruć ołowiem, cynkiem, talem, miedzią i złotem, cystynurii oraz reumatoidalnego zapalenia stawów, w którym inne metody leczenia okazały się nieskuteczne (poprawa kliniczna może nastąpić po 1-3 mies. stosowania penicylaminy). Przeciwwskazania: Nadwrażliwość na preparat lub penicylinę, niewydolność nerek, choroby tkanki łącznej z wyjątkiem reumatoidalnego zapalenia stawów, myasthenia gravis. Stosowanie penicylaminy jest przeciwwskazane u chorych, u których wystąpił białkomocz po leczeniu preparatami złota lub niedokrwistość aplastyczna czy agranulocytoza po uprzednim leczeniu penicylaminą. Interakcje: Penicylamina tworzy trwałe związki z wieloma metalami ciężkimi. Działanie niepożądane: Występują często i mogą mieć poważny przebieg, niekiedy prowadzący nawet do zgonu. Do najczęstszych należą reakcje alergiczne: świąd, osutka, pęcherzyca, gorączka, bóle stawów, powiększenie obwodowych węzłów chłonnych, toczeń polekowy, złuszczające zapalenie skóry, pokrzywka, wędrujące zapalenie stawów, zapalenie błony maziowej stawów, astma oskrzelowa; zapalenie gruczołu tarczowego; ze strony przewodu pokarmowego: zmniejszenie łaknienia, zapalenie języka z metalicznym posmakiem w ustach, dziąseł i błony śluzowej jamy ustnej, bóle brzucha, nudności, wymioty, biegunka, uczynnienie choroby wrzodowej żołądka, wewnątrzwątrobowy zastój żółci; objawy te ustępują po odstawieniu leku; hematologiczne: upośledzenie czynności szpiku kostnego, zwiększenie lub zmniejszenie liczby leukocytów i płytek krwi, eozynofilia, agranulocytoza, niedokrwistość aplastyczna, hemolityczna i niedoborowa; nerkowe: białkomocz, krwinkomocz, zespół nerczycowy, zespół Goodpasteure a; ze strony OUN: zapalenie nerwu wzrokowego, zaburzenia widzenia, szum w uszach, obwodowe neuropatie czuciowe i ruchowe, zaburzenia psychiczne; inne: myasthenia gravis, zakrzepowe zapalenie żył, wypadanie włosów, liszaj płaski, zapalenie wielomięśniowe, przerost gruczołu sutkowego, zapalenie oskrzeli, śródmiąższowe zapalenie płuc. Rozpad czynnika reumatoidalnego spowodowany penicylaminą może wywołać powstanie nowych przeciwciał, powodujących odmiedniczkowe zapalenie nerek, zapalenie mięśni lub zapalenie m. sercowego. Ciąża i laktacja: Nie stosować w ciąży (wyjątek choroba Wilsona - dawkę zmniejszyć do 1 g/d). Nie stosować w okresie karmienia piersią. Dawkowanie: Penicylaminę należy podawać 1 h przed posiłkiem lub 2 h po nim, zachowując przynajmniej godzinny odstęp po podaniu innych leków. Choroba Wilsona. Dawkowanie ustala się w zależności od stanu chorego i dobowej ilości miedzi wydalanej z moczem. Najczęściej rozpoczyna się od dobowej dawki 1 g, którą stopniowo zwiększa się do 2 g/d. Przewlekłe zatrucie ołowiem. Dorośli: 0,9-1,5 g/d w 3 dawkach podzielonych. Dzieci: mg/kg mc./d. Cystynuria. Leczenie rozpoczyna się od 250 mg/d., a następnie powoli zwiększa się dawkę. Dorośli: 2 g/d. Dzieci: mg/kg mc./d w 4 dawkach podzielonych. Reumatoidalne zapalenie stawów przez 1. mies. leczenia stosuje się 125 lub 250 mg/d; następnie dawkę zwiększa się co mies. o mg. Jeśli objawy kliniczne ustępują, leczenie kontynuuje się za pomocą najmniejszej skutecznej dawki. W razie utrzymywania się objawów klinicznych po 3 mies. stosowania dawki mg/d należy zwiększyć ją o kolejne 250 mg co mies. przez kolejne 3 mies. leczenia. Leczenie podtrzymujące: dawkowanie ustala się indywidualnie; najczęściej podaje się mg/d. Jeśli po 4-6 mies. leczenia penicylaminą w dawce max. 0,75 g/d nie występuje poprawa kliniczna, należy zrezygnować ze stosowania leku. Uwagi: Ze względu na częste występowanie działań niepożądanych i toksycznych penicylaminy zaleca się wykonywanie częstych badań kontrolnych morfologii krwi, jonogramu i badania ogólnego moczu - początkowo co 3 dni, następnie raz w tyg. W wypadku wystąpienia działań niepożądanych leczenie należy przerwać. Po ustąpieniu działań niepożądanych leczenie podejmuje się na nowo, rozpoczynając od najmniejszej dawki leku. Podczas leczenia za pomocą penicylaminy wskazane jest jednoczesne podawanie wit. B6. Jeśli preparaty złota okazały się nieskuteczne w leczeniu reumatoidalnego zapalenia stawów, leczenie penicylaminą można rozpocząć nie wcześniej niż 6 mies. po ich odstawieniu.")
72
baclofen myorelaxans M03BX
Działanie: Pochodna kwasu g-aminomasłowego (GABA) zmniejszająca napięcie mięśni szkieletowych. Hamuje odruchy mono- i polisynaptyczne na poziomie rdzenia kręgowego, najprawdopodobniej przez stymulację receptorów GABA-ergicznych (dokładny mechanizm działania nie został poznany). Wchłania się szybko i całkowicie po podaniu p.o. Maksymalne stężenie we krwi osiąga po 2 h. t1/2 we krwi wynosi 3-4 h. Z białkami krwi wiąże się w 30%. Metabolizowany w wątrobie w 10%. Wydala się prawie całkowicie przez nerki, głównie w postaci niezmienionej. Wskazania: W celu zmniejszenia napięcia mięśniowego w stwardnieniu rozsianym, po urazach i w chorobach rdzenia kręgowego. Nie jest skuteczny w stanach spastycznych pochodzenia mózgowego. Przeciwwskazania: Nadwrażliwość na lek, padaczka, stany drgawkowe, uszkodzenia mózgu i wiek do 12. rż. Ostrożnie w niewydolności nerek (należy zmniejszyć dawkę), chorobie wrzodowej, zaburzeniach psychicznych, zaburzeniach krążenia mózgowego i niewydolności oddechowej. Interakcje: Nasila działanie leków hipotensyjnych. W skojarzeniu z trójpierścieniowymi lekami przeciwdepresyjnymi znacznie zmniejsza napięcie mięśniowe i zaburza pamięć. Nasila działanie etanolu i leków działających depresyjnie na OUN. Działanie niepożądane: Senność, zawroty głowy, osłabienie, zmęczenie, splątanie, bóle głowy, bezsenność. Rzadziej inne zaburzenia neurologiczne i psychiczne. Niedociśnienie tętnicze, rzadziej duszność, kołatanie serca, ból w klatce piersiowej. Nudności, zaparcie, suchość błony śluzowej jamy ustnej, bóle brzucha, wymioty, biegunka, częstomocz, dolegliwości dysuryczne, zatrzymanie moczu, impotencja, krwiomocz, rumień, świąd, obrzęk podudzi, zwiększenie aktywności enzymów wątrobowych i stężenia glukozy we krwi. Objawy przedawkowania: wymioty, zmniejszone napięcie mięśniowe, senność, zaburzenia akomodacji, śpiączka, depresja oddechowa, drgawki. Postępowanie: opróżnić żołądek (prowokacja wymiotów lub płukanie) i zabezpieczyć drogi oddechowe. Ciąża i laktacja: Kategoria C. W okresie karmienia piersią stosować tylko w wyjątkowych przypadkach (przenika do pokarmu kobiecego). Dawkowanie: Dawka powinna być zindywidualizowana. Początkowo 5 mg 3 razy na dobę, następnie zwiększać dawkę dobową co 3 dni o 15 mg do uzyskania pożądanego efektu. Nie przekraczać dawki 80 mg/d (w 3-4 dawkach podzielonych). Zmniejszyć dawkę w niewydolności nerek. Może wystąpić zmniejszenie skuteczności leku po długotrwałym stosowaniu. Po nagłym odstawieniu może wystąpić powrót objawów w większym nasileniu (efekt "odbicia") - redukować dawkę leku stopniowo w ciągu kilku dni. Uwagi: Upośledza zdolność kierowania pojazdami i obsługę urządzeń mechanicznych. Preparaty baclofen
 zmniejszająca napięcie mięśni szkieletowych. Hamuje odruchy mono- i polisynaptyczne na poziomie rdzenia kręgowego, najprawdopodobniej przez stymulację receptorów GABA-ergicznych (dokładny mechanizm działania nie został poznany). Wchłania się szybko i całkowicie po podaniu p.o. Maksymalne stężenie we krwi osiąga po 2 h. t1/2 we krwi wynosi 3-4 h. Z białkami krwi wiąże się w 30%. Metabolizowany w wątrobie w 10%. Wydala się prawie całkowicie przez nerki, głównie w postaci niezmienionej. Wskazania: W celu zmniejszenia napięcia mięśniowego w stwardnieniu rozsianym, po urazach i w chorobach rdzenia kręgowego. Nie jest skuteczny w stanach spastycznych pochodzenia mózgowego. Przeciwwskazania: Nadwrażliwość na lek, padaczka, stany drgawkowe, uszkodzenia mózgu i wiek do 12. rż. Ostrożnie w niewydolności nerek (należy zmniejszyć dawkę), chorobie wrzodowej, zaburzeniach psychicznych, zaburzeniach krążenia mózgowego i niewydolności oddechowej. Interakcje: Nasila działanie leków hipotensyjnych. W skojarzeniu z trójpierścieniowymi lekami przeciwdepresyjnymi znacznie zmniejsza napięcie mięśniowe i zaburza pamięć. Nasila działanie etanolu i leków działających depresyjnie na OUN. Działanie niepożądane: Senność, zawroty głowy, osłabienie, zmęczenie, splątanie, bóle głowy, bezsenność. Rzadziej inne zaburzenia neurologiczne i psychiczne. Niedociśnienie tętnicze, rzadziej duszność, kołatanie serca, ból w klatce piersiowej. Nudności, zaparcie, suchość błony śluzowej jamy ustnej, bóle brzucha, wymioty, biegunka, częstomocz, dolegliwości dysuryczne, zatrzymanie moczu, impotencja, krwiomocz, rumień, świąd, obrzęk podudzi, zwiększenie aktywności enzymów wątrobowych i stężenia glukozy we krwi. Objawy przedawkowania: wymioty, zmniejszone napięcie mięśniowe, senność, zaburzenia akomodacji, śpiączka, depresja oddechowa, drgawki. Postępowanie: opróżnić żołądek (prowokacja wymiotów lub płukanie) i zabezpieczyć drogi oddechowe. Ciąża i laktacja: Kategoria C. W okresie karmienia piersią stosować tylko w wyjątkowych przypadkach (przenika do pokarmu kobiecego). Dawkowanie: Dawka powinna być zindywidualizowana. Początkowo 5 mg 3 razy na dobę, następnie zwiększać dawkę dobową co 3 dni o 15 mg do uzyskania pożądanego efektu. Nie przekraczać dawki 80 mg/d (w 3-4 dawkach podzielonych). Zmniejszyć dawkę w niewydolności nerek. Może wystąpić zmniejszenie skuteczności leku po długotrwałym stosowaniu. Po nagłym odstawieniu może wystąpić powrót objawów w większym nasileniu (efekt odbicia ) - redukować dawkę leku stopniowo w ciągu kilku dni. Uwagi: Upośledza zdolność kierowania pojazdami i obsługę urządzeń mechanicznych. Preparaty baclofen.")
73
Interferony grupa białek wydzielanych przez komórki głównie w odpowiedzi na zakażenie wirusowe. Interferony przeszkadzają wirusom w atakowaniu kolejnych komórek, bo między innymi zwiększają produkcję białek MHC klasy I (i w ten sposób ułatwiają cytotoksycznym limfocytom T niszczenie komórek zainfekowanych wirusami) oraz pobudzają geny hamujące metabolizm wirusów. Wyróżnia się trzy główne grupy interferonów. Interferon alfa jest wytwarzany głównie przez różne leukocyty (między innymi makrofagi, limfocyty B i limfocyty NK); interferon beta - przez fibroblasty, a interferon gamma - przez limfocyty. Interferony hamują także mnożenie się komórek nowotworowych. Sztucznie uzyskany interferon stosuje się w leczeniu niektórych chorób nowotworowych (np. niektórych białaczek i raka nerki) oraz stwardnienia rozsianego.
 oraz pobudzają geny hamujące metabolizm wirusów. Wyróżnia się trzy główne grupy interferonów. Interferon alfa jest wytwarzany głównie przez różne leukocyty (między innymi makrofagi, limfocyty B i limfocyty NK); interferon beta - przez fibroblasty, a interferon gamma - przez limfocyty. Interferony hamują także mnożenie się komórek nowotworowych. Sztucznie uzyskany interferon stosuje się w leczeniu niektórych chorób nowotworowych (np. niektórych białaczek i raka nerki) oraz stwardnienia rozsianego..")
74
Drżenie pęczkowe : powstaje wskutek szybkich skurczów włókienek mięśniowych jest wyrazem patologicznych wyładowań ze schorzałej kom. ruchowej są to niewielkie drgania podskórne odpowiadające skurczom jednostkiruchowej najcz. w przewlekłych sprawach zwyrodnieniowych uszkadazjącychkom. ruchowe rogów przednich rdzenia np. stwardnienie boczne zanikowe

75
chód paretyczny (niedowładny) :
BADANIE CHODU : zwracamy uwagę na kończyny górne i dolne, na postawę chorego podczas chodzenia chory najpierw chodzi z otwartymi, a potem z zamkniętymi oczami chód paretyczny (niedowładny) : chory chodzi powoli, małymi krokami, z trudnością przesuwa stopy (powłuczy nogami) jeśli niedowład jest szczególnie nasilony w prostownikach stóp chód staje się brodzący występowanie : niedowład wiotki kończyn dolnych chód brodzący : z powodu opadnięcia stóp chory nadmiernie zgina nogi w kolanach zapalenie wielonerwowe chód kaczkowaty : podczas chodzenia kołysanie w biodrach uderzająca jest trudność wchodzenia na schody występuje niedowład mm. obręczy i ud postępująca dystrofia mięśniowa (dystrophia musculorum progressiva)
 : chory chodzi powoli, małymi krokami, z trudnością przesuwa stopy (powłuczy nogami) jeśli niedowład jest szczególnie nasilony w prostownikach stóp chód staje się brodzący. występowanie : niedowład wiotki kończyn dolnych. chód brodzący : z powodu opadnięcia stóp chory nadmiernie zgina nogi w kolanach. zapalenie wielonerwowe. chód kaczkowaty : podczas chodzenia kołysanie w biodrach. uderzająca jest trudność wchodzenia na schody. występuje niedowład mm. obręczy i ud. postępująca dystrofia mięśniowa (dystrophia musculorum progressiva)")
76
Drżenie włókienkowe (w obrębie jezyka):
występuje w odnerwionych włóknach mięśniowych powstaje na skutek nadwrażliwości wyrodniejących włókien mięśniowych w stosunku do Ach tkankowej nie jest widoczne pod skórą

77
riluzole neuroprotectivum N07XX Działanie: Pochodna benzotiazolu, hamuje uwalnianie glutaminianu, inaktywuje zależne od napięcia kanały sodowe, ma wpływ na procesy wewnątrzkomórkowe będące efektem połączenia neuroprzekaźników z receptorami aminokwasów. Wykazano, że lek hamuje presynaptyczne uwalnianie aminokwasów pobudzających: asparaginianu i glutaminianu (w stężeniach mikromolarnych), a także kwasu g-aminomasłowego (GABA) i glicyny. Hamuje wychwyt neuronalny dopaminy, GABA i glutaminianu, nie wpływa na wychwyt asparaginianu. Zmniejsza efekty będące następstwem stymulacji receptorów dla aminokwasów pobudzających. Nie wiadomo dokładnie, który z powyższych mechanizmów odpowiada za efekty, jakie riluzol wywiera w trakcie leczenia stwardnienia zanikowego bocznego. W wielu doświadczalnych modelach uszkodzenia komórek nerwowych in vivo riluzol wykazywał także działanie neuroprotekcyjne. Poprzez hamowanie glutaminianu riluzol powoduje rozluźnienie mięśni i sedację. Wykazuje również właściwości przeciwdrgawkowe. Dobrze wchłania się po podaniu p.o., jego dostępność biologiczna wynosi ok. 60%. Tłuszcze zmniejszają wchłanianie riluzolu. t1/2 wynosi 9-15 h. W 97% wiąże się z białkami osocza. Ulega kumulacji, stan stacjonarny osiąga w ciągu 5 dni. Metabolizm riluzolu następuje w wątrobie w układzie cytochromu P-450; jest on sprzęgany z kwasem glukuronowym. Wydalany głównie z moczem, częściowo z żółcią. Niewydolność wątroby i(lub) nerek upośledza wydalanie i powoduje zwiększenie jego stężenia w surowicy. Palenie tytoniu powoduje szybszą eliminację leku z ustroju; jest szybciej metabolizowany u mężczyzn. Wskazania: Leczenie pacjentów ze stwardnieniem zanikowym bocznym - wydłuża przeżycie i(lub) opóźnia wystąpienie niewydolności oddechowej. Nie likwiduje objawów, które wystąpiły przed podaniem leku, częściowo hamuje dalszy rozwój choroby. W badaniach klinicznych z grupą kontrolną stwierdzono wydłużenie średniego przeżycia o ok. 90 dni. Przeciwwskazania: Nadwrażliwość na lek, istotne upośledzenie czynności nerek lub wątroby. Ostrożnie u chorych w wieku podeszłym. Podczas leczenia należy oznaczać aktywność aminotransferaz, przez pierwsze 3 miesiące co miesiąc, następnie co 3 miesiące, po roku okresowo. W przypadku 5-krotnego zwiększenia ich aktywności w stosunku do górnej granicy normy należy zaprzestać leczenia. Nie ustalono skuteczności i bezpieczeństwa stosowania u dzieci. Interakcje: W przeprowadzonych badaniach nie stwierdzono istotnych interakcji z innymi lekami. Ponieważ riluzol jest metabolizowany przez CYP1A2, inhibitory tego izoenzymu mogą nasilać jego metabolim. Działanie niepożądane: Osłabienie, nudności, wymioty, zawroty głowy, biegunka, zaparcia, bóle brzucha, zapalenie płuc, parestezje wokół ust, brak łaknienia, senność, zwiększenie aktywności enzymów wątrobowych. Rzadko zaburzenia oddechowe, trudności w połykaniu, objawy grypopodobne, zatrzymanie czynności serca, zapalenie oskrzeli. Bardzo rzadko reakcje rzekomoanafilaktyczne, obrzęk naczynioruchowy, zapalenie trzustki. W przypadku przedawkowania występuje methemoglobinemia; leczenie objawowe i podtrzymujące. Ciąża i laktacja: Kategoria C. Nie stosować w ciąży i w okresie karmienia piersią. Dawkowanie: 50 mg co 12 h p.o. 1 h przed posiłkiem lub 2 h po nim. Uwagi: Podczas stosowania leku może wystąpić osłabienie koncentracji i zdolności do prowadzenia pojazdów lub obsługi urządzeń w ruchu. Preparaty riluzole
 opóźnia wystąpienie niewydolności oddechowej. Nie likwiduje objawów, które wystąpiły przed podaniem leku, częściowo hamuje dalszy rozwój choroby. W badaniach klinicznych z grupą kontrolną stwierdzono wydłużenie średniego przeżycia o ok. 90 dni. Przeciwwskazania: Nadwrażliwość na lek, istotne upośledzenie czynności nerek lub wątroby. Ostrożnie u chorych w wieku podeszłym. Podczas leczenia należy oznaczać aktywność aminotransferaz, przez pierwsze 3 miesiące co miesiąc, następnie co 3 miesiące, po roku okresowo. W przypadku 5-krotnego zwiększenia ich aktywności w stosunku do górnej granicy normy należy zaprzestać leczenia. Nie ustalono skuteczności i bezpieczeństwa stosowania u dzieci. Interakcje: W przeprowadzonych badaniach nie stwierdzono istotnych interakcji z innymi lekami. Ponieważ riluzol jest metabolizowany przez CYP1A2, inhibitory tego izoenzymu mogą nasilać jego metabolim. Działanie niepożądane: Osłabienie, nudności, wymioty, zawroty głowy, biegunka, zaparcia, bóle brzucha, zapalenie płuc, parestezje wokół ust, brak łaknienia, senność, zwiększenie aktywności enzymów wątrobowych. Rzadko zaburzenia oddechowe, trudności w połykaniu, objawy grypopodobne, zatrzymanie czynności serca, zapalenie oskrzeli. Bardzo rzadko reakcje rzekomoanafilaktyczne, obrzęk naczynioruchowy, zapalenie trzustki. W przypadku przedawkowania występuje methemoglobinemia; leczenie objawowe i podtrzymujące. Ciąża i laktacja: Kategoria C. Nie stosować w ciąży i w okresie karmienia piersią. Dawkowanie: 50 mg co 12 h p.o. 1 h przed posiłkiem lub 2 h po nim. Uwagi: Podczas stosowania leku może wystąpić osłabienie koncentracji i zdolności do prowadzenia pojazdów lub obsługi urządzeń w ruchu. Preparaty riluzole.")
78
Leczenie antycholinergiki Atropina , doustnie, 0.6 mg x4/24h
Propantelina ,doustnie, 15mg 3x/24h do dawki 30 mg 4x/24h Hioscyna, plaster naklejany na skórę za uchem zapewnia powolne uwalnianie leku przez 72h Inne ważne : W razie dławienia przechylenie pacjenta do przodu, odessanie substancji wydzielniczej lub pokarmowej z drzewa oskrzelowego U chorych z problemami z bezsennoscią przepisac leczenie farmakologiczne Stosuje się odpowiednie szyny w celu podtrzymania stawów, oraz kołnierz podtrzymujący szyję W przypadkach silnego kurczu mieśniowego gdy powoduje to ból wskazane jest leczenie farmakologiczne bólu i fizykoterapia Wazna jest odpowiednie przystosowanie mieszkania chorego oraz zapewnienie niezbednej rehabilitacji ruchowej W koncowej fazie choroby należy zadbac o czeste zmiany pozycji chorego, odpowiednie materace , łóżka Należy zwracać uwagę na odpowiednia konsystencję i wartość odżywczą posiłków. Spadek masy ciała jest nieunikniony, bywa często alarmujący dla chorego.Pokarmy płynne sprawiają mniej rudności niż papkowate W przypadku spastyczności mięśni gardła przydatne jest ssanie kostki lodu, lub przykładanie okładu z lodu na gardło przed posiłkiem Pomocne lub nawet niezbedne może okazac się uzywanie zgłębnika lub wykonanie gastrostomii W przypadkach bólów konczyn stosuje się NLPZ, a w końcowych fazach choroby morfinę(5-10mg doustnie – w razie potrzeby) W koncowym okresie choroby konieczne staje się zapewnienie właściwej opieki w szpitalu lub hospicjum Wazne jest zapewnienie chorym towazystwa innych osób
 W koncowym okresie choroby konieczne staje się zapewnienie właściwej opieki w szpitalu lub hospicjum. Wazne jest zapewnienie chorym towazystwa innych osób.")
79
Przyczyna/uszkodzenie
Leczenie/techniki Narastający niedowład mięśnia przepoby powodujacy niewydolność oddechową Intubacja, tracheostomia, oddech kontrolowany Porazenie opuszki Zgłębnik dożołądkowy, opieka na oddziale intensywnej terapii, gastrostomia Infekcje ukł.oddechowego profilaktyka antybiotykowa, fizykoterapia klatki piersiowej, czynne odsysanie zalegającej wydzieliny drzewa oskrzelowego odleżyny Fizykoterapia, zabiegi chirurgiczne Aparat mowy Tablice do komunikacji, wzglednie inne techniki komunikacyjne – zależne od stopnia ciężkości choroby, np.. syntezatory mowy, komputery, sterowanie kursora ruchem gałek ocznych, predefiniowane słowniki Zaleganie moczu Cewnik do pęcherza, antybiotykoterapia i profilaktyka antybiotykowa

80
Problemy na jakie napotykaja naukowcy badając ALS zarówno w zakresie nauk klinicznych jak i podstawowych Badania prowadzone sa niekiedy na bardzo małym materiale (brak grupy kontrolnej, liczne niecałkowicie uzasadnione koncepcje etiopatogenetyczne) Mnogość szczegółowych badań i hipotez nastrecza trudności w ich klasyfikacji Od 1869 roku tj. od opisania przez Charcot’a i Joffroy’a dzieki licznym badaniom zarejestrowano jedynie liczne szczegółowe „znaleziska” mające znaczenie poznawcze, ale nie udało się pomimo wysiłku poszerzyc wiedzy dotyczacej czynnika lub czynników sprawchych ALS
 Mnogość szczegółowych badań i hipotez nastrecza trudności w ich klasyfikacji. Od 1869 roku tj. od opisania przez Charcot’a i Joffroy’a dzieki licznym badaniom zarejestrowano jedynie liczne szczegółowe „znaleziska mające znaczenie poznawcze, ale nie udało się pomimo wysiłku poszerzyc wiedzy dotyczacej czynnika lub czynników sprawchych ALS.")
81
Professor Stephen Hawking

82
Przypadek kliniczny Professor Hawking supplements his diet with daily mineral and vitamin tablets, and zinc, cod liver oil capsules, folic acid, vitamin B complex, vitamin B-12, vitamin C and vitamin E are said to have been particularly helpful. He also follows a diet free of gluten and vegetable oil and avoids convenience foods; quite recently he started to include a small amount of dairy produce. As far as medical care is concerned, he receives passive chest physiotherapy and passive and active physiotherapy to all limbs and muscle groups. He is not currently undergoing any new treatment, and does not have a particular specialist to look after him. He does have his own GP. In a note on his experiences with MND, which appears on his own website ( org.uk), he says: "It was a great shock to me to discover that I had motor neurone disease. In my third year at Oxford, I noticed that I seemed to be getting more clumsy, and I fell over once or twice for no apparent reason. But it was not until I was at Cambridge that my father noticed, and took me to the family doctor. He referred me to a specialist, and shortly after my 21 st birthday, I went into hospital for tests. "I was in for two weeks, during which I had a wide variety of tests. After all that, they didn't tell me what I had, except that it was not multiple sclerosis, and that I was an atypical case. I gathered, however, that they expected it to continue to get worse, and that there was nothing they could do, except give me vitamins. I could see that they didn't expect them to have much effect. I didn't feel like asking for more details, because they were obviously bad. "The realisation that I had an incurable disease that was likely to kill me in a few years was a bit of a shock." He later married Jane Wilde and they had three children. His condition gradually deteriorated but he managed to cope with the help of his wife and research students until 1980 when he changed to a system of community and private nurses. "This lasted until I caught pneumonia in I had to have a tracheostomy operation. After this, I had to have 24 hour nursing care, made possible by grants from several foundations... The tracheostomy operation removed my ability to speak altogether. "For a time, the only way I could communicate was to spell out words letter by letter, by raising my eyebrows when someone pointed to the right letter on a spelling card. It is pretty difficult to carry on a conversation like that, let alone write a scientific paper." "A computer expert in California, Walt Woltosz, heard of my plight. He sent me a computer program he had written. This allowed me to select words from a series of menus on the screen, by pressing a switch in my hand. The program could also be controlled by a switch, operated by head or eye movement. When I have built up what I want to say, I can send it to a speech synthesiser. "I have had motor neurone disease for practically all my adult life. Yet it has not prevented me from having a very attractive family, and being successful in my work. This is thanks to the help I have received from Jane, my children, and a large number of other people and organisations. I have been lucky, that my condition has progressed more slowly than is often the case. But it shows that one need not lose hope."
, he says: It was a great shock to me to discover that I had motor neurone disease. In my third year at Oxford, I noticed that I seemed to be getting more clumsy, and I fell over once or twice for no apparent reason. But it was not until I was at Cambridge that my father noticed, and took me to the family doctor. He referred me to a specialist, and shortly after my 21 st birthday, I went into hospital for tests. I was in for two weeks, during which I had a wide variety of tests. After all that, they didn t tell me what I had, except that it was not multiple sclerosis, and that I was an atypical case. I gathered, however, that they expected it to continue to get worse, and that there was nothing they could do, except give me vitamins. I could see that they didn t expect them to have much effect. I didn t feel like asking for more details, because they were obviously bad. The realisation that I had an incurable disease that was likely to kill me in a few years was a bit of a shock. He later married Jane Wilde and they had three children. His condition gradually deteriorated but he managed to cope with the help of his wife and research students until 1980 when he changed to a system of community and private nurses. This lasted until I caught pneumonia in I had to have a tracheostomy operation. After this, I had to have 24 hour nursing care, made possible by grants from several foundations... The tracheostomy operation removed my ability to speak altogether. For a time, the only way I could communicate was to spell out words letter by letter, by raising my eyebrows when someone pointed to the right letter on a spelling card. It is pretty difficult to carry on a conversation like that, let alone write a scientific paper. A computer expert in California, Walt Woltosz, heard of my plight. He sent me a computer program he had written. This allowed me to select words from a series of menus on the screen, by pressing a switch in my hand. The program could also be controlled by a switch, operated by head or eye movement. When I have built up what I want to say, I can send it to a speech synthesiser. I have had motor neurone disease for practically all my adult life. Yet it has not prevented me from having a very attractive family, and being successful in my work. This is thanks to the help I have received from Jane, my children, and a large number of other people and organisations. I have been lucky, that my condition has progressed more slowly than is often the case. But it shows that one need not lose hope.")
83
Przypadek kliniczny Stephen Hawking has survived almost 40 years with a disease that usually kills people 14 months after diagnosis. Roger Dobson asks why : Stephen Hawking believes motor neurone disease is a syndrome that can have different causes. "Maybe my variety is due to bad absorption of vitamins" Stephen Hawking developed motor neurone disease when he was in his early 20s. Most patients with the condition die within five years, and according to the Motor Neurone Disease Association, average life expectancy after diagnosis is 14 months. But Professor Hawking, the Cambridge University physicist and cosmologist and author of A Brief History of Time, has confounded the statistics and recently celebrated his 60th birthday. No one is thought to have survived for so long with the incurable condition, which kills three people a day in the United Kingdom. "Stephen Hawking is a fascinating case, and neurologists always puzzle over it. The case is fascinating because of the early onset and the length of time the disease has run," one neurologist said. Motor neurone disease (MND), or amyotrophic lateral sclerosis, is a progressive, usually fatal neuromuscular disease. It attacks motor neurones in the spinal cord and lower brain, which transmit signals from the brain to the voluntary muscles throughout the body. "The average duration of survival from diagnosis is about 14 months, but it varies enormously," says Professor Nigel Leigh, professor of clinical neurology at King's College, London, and director of the King's MND Care and Research Centre. "We have found that the surrival in younger patients is strikingly better and is measured in many years-in some cases more than 10. Among people in their 50s and 60s, there is a 50% chance of surviving four years or so. It is a different beast if you start young, oddly, and no one knows why. But even some forms of MND that start in the [patient's] 50s or 60s can be slowly progressive," he added. "I have no personal knowledge of Stephen Hawking, but he is exceptional. I am not aware of anyone else who has survived with MND as long. What is unusual is not only the length of time, but that the disease seems to have almost burnt out. He appears to be relatively stable, and I have had one or two patients where there is still a gradual deterioration, but where the curve has flattened off. In these cases MND started quite early, in the [patient's] 20s or 30s. This kind of stabilisation is extremely rare. "In early onset patients there appear to be biological differences. It is already clear that if you look at the genetics of MND, there are at least half a dozen, maybe a dozen, genetic forms of true MND. Another possibility is that there is some kind of interaction with the ageing process." Professor Pam Shaw, professor of neurology at the University of Sheffield, said: "The older you are the quicker the disease course tends to be, but we don't really have a handle on why some people survive for longer periods than others. I wish we did." Asked by the BMJ if he knew why his condition had evolved differently from a typical case of MND, Professor Hawking replied, "I believe motor neurone disease is a syndrome that can have different causes. Maybe my variety is due to bad absorption of vitamins."
, or amyotrophic lateral sclerosis, is a progressive, usually fatal neuromuscular disease. It attacks motor neurones in the spinal cord and lower brain, which transmit signals from the brain to the voluntary muscles throughout the body. The average duration of survival from diagnosis is about 14 months, but it varies enormously, says Professor Nigel Leigh, professor of clinical neurology at King s College, London, and director of the King s MND Care and Research Centre. We have found that the surrival in younger patients is strikingly better and is measured in many years-in some cases more than 10. Among people in their 50s and 60s, there is a 50% chance of surviving four years or so. It is a different beast if you start young, oddly, and no one knows why. But even some forms of MND that start in the [patient s] 50s or 60s can be slowly progressive, he added. I have no personal knowledge of Stephen Hawking, but he is exceptional. I am not aware of anyone else who has survived with MND as long. What is unusual is not only the length of time, but that the disease seems to have almost burnt out. He appears to be relatively stable, and I have had one or two patients where there is still a gradual deterioration, but where the curve has flattened off. In these cases MND started quite early, in the [patient s] 20s or 30s. This kind of stabilisation is extremely rare. In early onset patients there appear to be biological differences. It is already clear that if you look at the genetics of MND, there are at least half a dozen, maybe a dozen, genetic forms of true MND. Another possibility is that there is some kind of interaction with the ageing process. Professor Pam Shaw, professor of neurology at the University of Sheffield, said: The older you are the quicker the disease course tends to be, but we don t really have a handle on why some people survive for longer periods than others. I wish we did. Asked by the BMJ if he knew why his condition had evolved differently from a typical case of MND, Professor Hawking replied, I believe motor neurone disease is a syndrome that can have different causes. Maybe my variety is due to bad absorption of vitamins.")
Podobne prezentacje


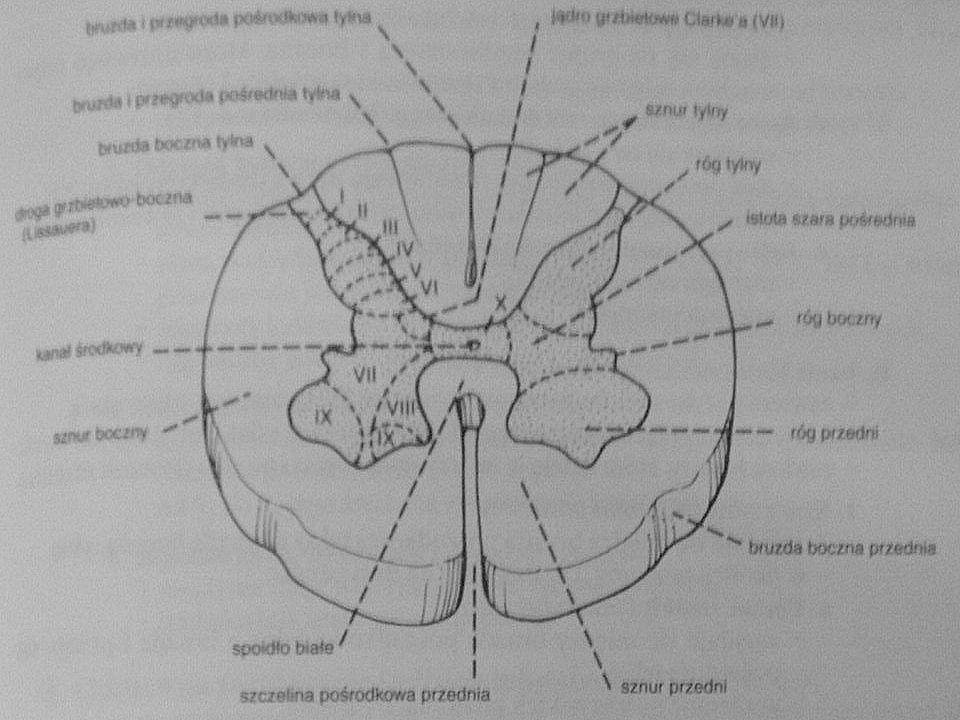
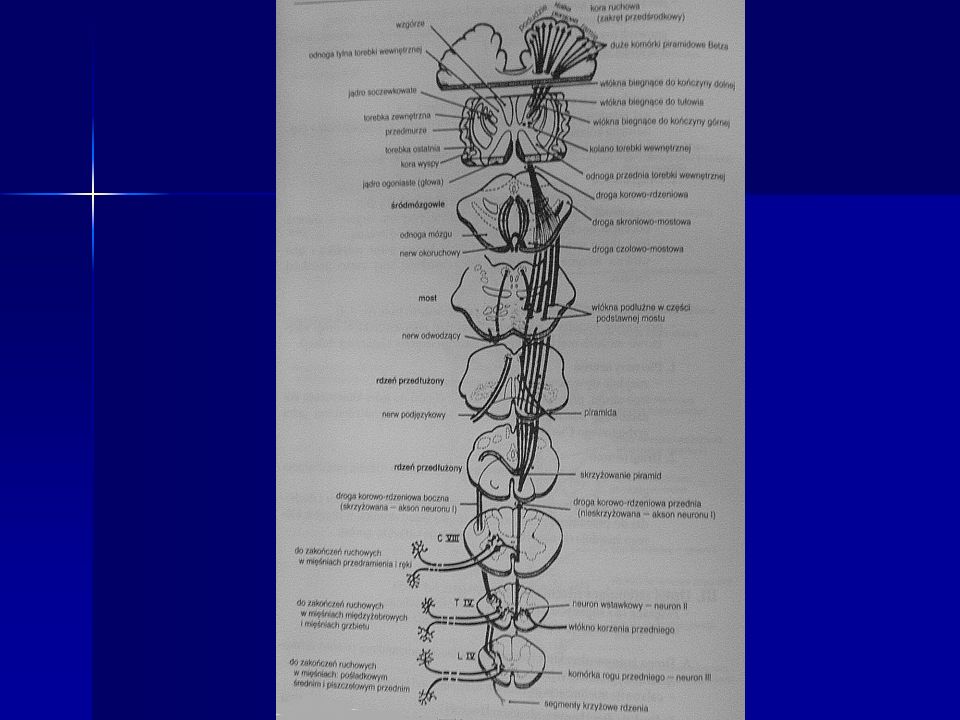
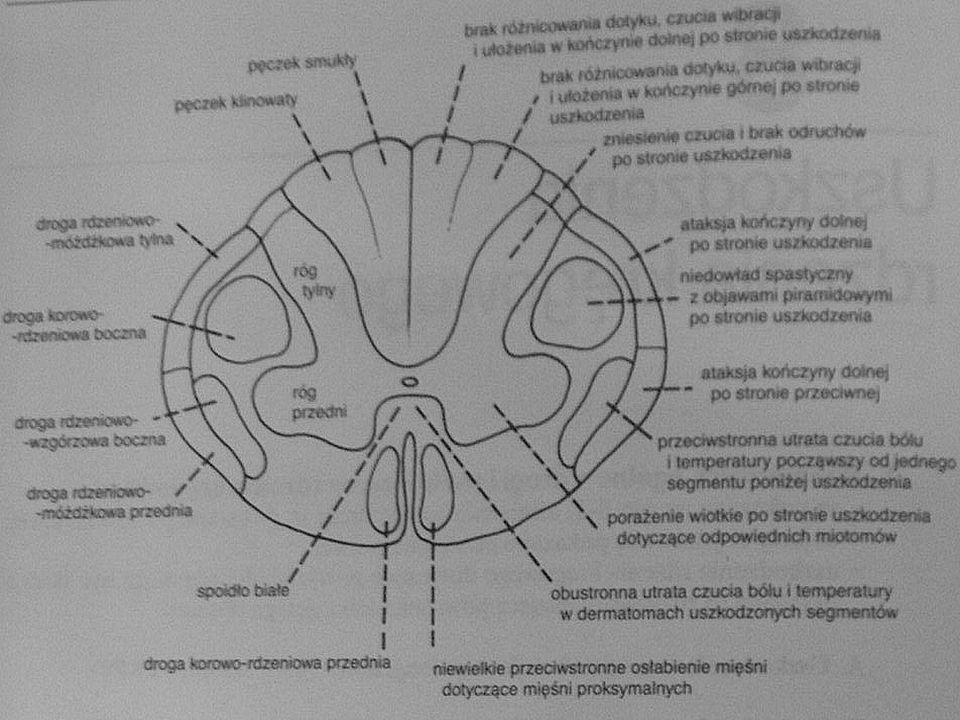
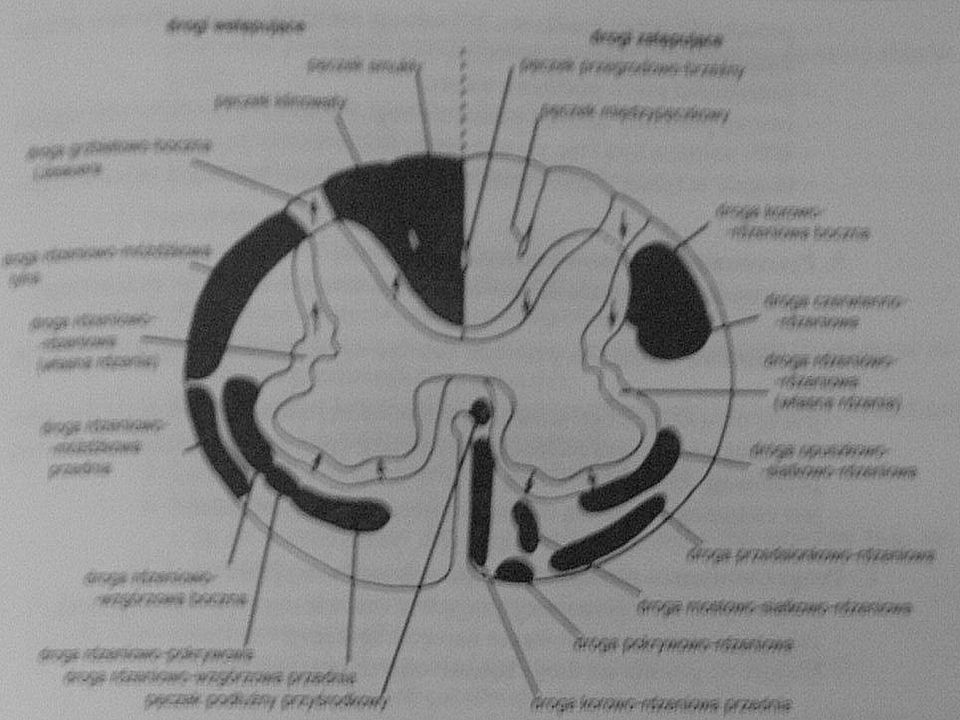
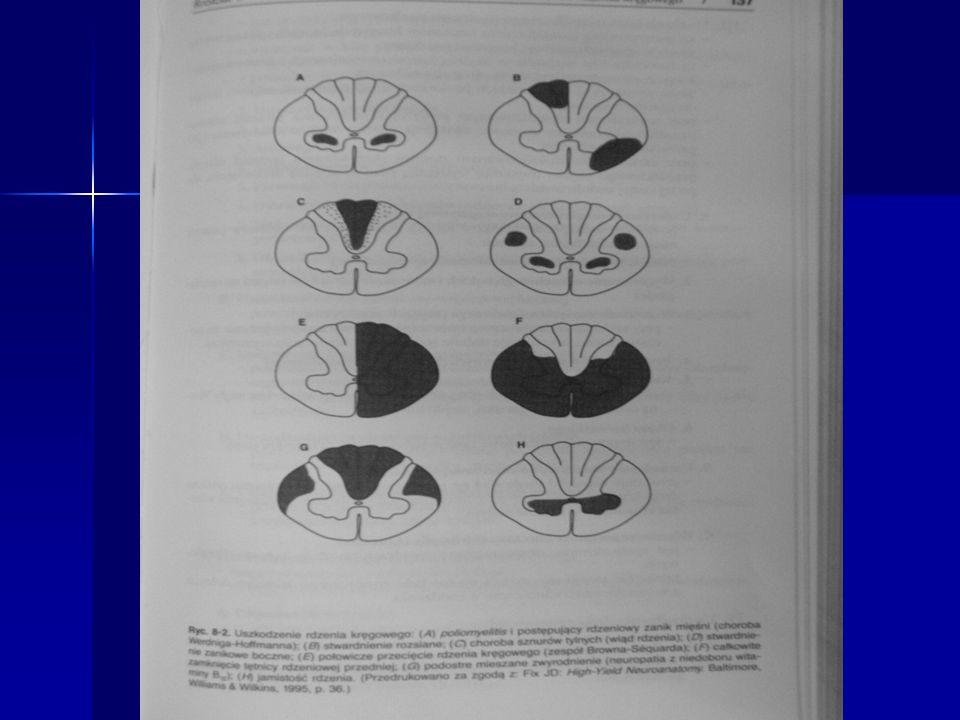












 – współczynnik powstały przez podzielenie masy.>")




 wynikającą z defektu produkcji lub działania insuliny wydzielanej.>")


