Pobierz prezentację
1
ZAKRES OPRACOWANIA I. FIZYKOCHEMICZNE WŁASNOŚCI PROCESU PALENIA
CIAŁ STAŁYCH Mechanizm spalania się ciał stałych w zależności od ich budowy wewnętrznej Chemiczna budowa drewna Fizyczna budowa drewna Rozprzestrzenianie się płomienia na powierzchni i szybkość liniowa palenia się drewna Czynniki wpływające na proces palenia się drewna: wilgotność, twardość, stosunek ilości do objętości, temperatura początkowa. Palenie się ciał strzępiastych Obserwacja faz palenia się drewna Określenie temperatury zapalenia

2
II. FIZYKOCHEMICZNE WŁASNOŚCI CIECZY PALNYCH
Parowanie cieczy Para nasycona i nienasycona Ciśnienie pary nasyconej Stężenie par nasyconych Szybkość parowania Temperatura zapłonu cieczy jednorodnych i niejednorodnych Klasyfikacja cieczy palnych Mechanizm zapalenia i palenia się cieczy palnych Mechanizm spalania cieczy Szybkość spalania – szybkość wagowa i liniowa Przegrzewanie się cieczy palnych w procesie palenia Wrzenie i wyrzut ze zbiorników podczas palenia

3
WŁASNOŚCI WYBUCHOWE PALNYCH GAZÓW
Przebieg procesu palenia gazów i par Palenie dyfuzyjne i kinetyczne Granice wybuchowości Bodziec termiczny Domieszki gazów obojętnych i par Temperatura mieszaniny wybuchowej Kierunek rozprzestrzeniania się płomienia w mieszaninach gazowych Klasyfikacja pożarowo – wybuchowa gazów i par cieczy palnych PODSUMOWANIE PRACY Materiały stałe Ciecze Gazy

4
FIZYKOCHEMICZNE WŁASNOŚCI CIECZY PALNYCH

5
Parowanie cieczy Parowanie definiowane jest jako przejście cząsteczek znajdujących się na powierzchni cieczy (stanu ciekłego) w stan gazowy, natomiast wrzenie jest określane jako parowanie z całej objętości cieczy. Parowanie cieczy zachodzi w każdej temperaturze, natomiast wrzenie w temperaturze nazywanej temperaturą wrzenia. Rozpatrując molekularny model cieczy można łatwo zrozumieć te zjawiska, oraz przekonać się, że definicja wrzenia nie jest w pełni poprawna. Załóżmy, że nad cieczą znajdującą się w zamkniętym nieważkim tłokiem naczyniu znajduje się powietrze. Cząsteczki cieczy poruszają się chaotycznie i zderzają ze sobą (praktycznie są to zderzenia sprężyste). Od czasu do czasu cząsteczka znajdująca się na powierzchni cieczy zostaje wyrzucona do fazy gazowej. W fazie gazowej nad cieczą ciśnienie musi być równe ciśnieniu atmosferycznemu. Gdy w fazie gazowej znajdą się cząsteczki pochodzące od cieczy (para), to sumaryczna ilość tych cząsteczek zwiększy się (pary cieczy i powietrze) i wtedy zauważymy, że tłok przesunie się ku górze, co jest zgodne z prawem Daltona p = pc+pp. Gdy zwiększa się prężność par pc to prężność powietrza pp musi zmaleć, by ciśnienie całkowite (p = const), było stałe . Prężność powietrza może zmaleć wtedy, gdy zwiększy się objętość nad cieczą (PV = const). Na tłok poruszający i przesuwający się ku górze działa siła równa ciśnieniu atmosferycznemu. Układ wykonuje więc pewną pracę, na którą potrzebna jest mu energia. Dodatkowa energia potrzebna jest na pokonanie sił wzajemnego przyciągania się cząsteczek cieczy (sił spójności).
 w stan gazowy, natomiast wrzenie jest określane jako parowanie z całej objętości cieczy. Parowanie cieczy zachodzi w każdej temperaturze, natomiast wrzenie w temperaturze nazywanej temperaturą wrzenia. Rozpatrując molekularny model cieczy można łatwo zrozumieć te zjawiska, oraz przekonać się, że definicja wrzenia nie jest w pełni poprawna. Załóżmy, że nad cieczą znajdującą się w zamkniętym nieważkim tłokiem naczyniu znajduje się powietrze. Cząsteczki cieczy poruszają się chaotycznie i zderzają ze sobą (praktycznie są to zderzenia sprężyste). Od czasu do czasu cząsteczka znajdująca się na powierzchni cieczy zostaje wyrzucona do fazy gazowej. W fazie gazowej nad cieczą ciśnienie musi być równe ciśnieniu atmosferycznemu. Gdy w fazie gazowej znajdą się cząsteczki pochodzące od cieczy (para), to sumaryczna ilość tych cząsteczek zwiększy się (pary cieczy i powietrze) i wtedy zauważymy, że tłok przesunie się ku górze, co jest zgodne z prawem Daltona p = pc+pp. Gdy zwiększa się prężność par pc to prężność powietrza pp musi zmaleć, by ciśnienie całkowite (p = const), było stałe . Prężność powietrza może zmaleć wtedy, gdy zwiększy się objętość nad cieczą (PV = const). Na tłok poruszający i przesuwający się ku górze działa siła równa ciśnieniu atmosferycznemu. Układ wykonuje więc pewną pracę, na którą potrzebna jest mu energia. Dodatkowa energia potrzebna jest na pokonanie sił wzajemnego przyciągania się cząsteczek cieczy (sił spójności).")
6
Część energii wewnętrznej układu zostaje zamieniona na pracę objętościową (przesunięcie tłoka) i pokonanie sił spójności. Dlatego ciecz ulega ochłodzeniu. Do identycznego wniosku dojdziemy rozpatrując energię cząsteczek w stanie ciekłym. Średnia energia a co zatem idzie i średnia prędkość jest proporcjonalna do temperatury. Im wyższa temperatura, tym wyższa prędkość cząsteczek cieczy, ponieważ tylko cząsteczki o odpowiednio dużej prędkości (energii) mogą przejść w stan pary. Gdy w fazie ciekłej ubywa cząsteczek o dużej energii obniża się średnia energia cząsteczek, czyli obniża się temperatura układu. Po pewnym czasie w stanie gazowym znajdzie się tak duża ilość cząsteczek pochodzących od cieczy, że każda następna cząsteczka przechodząca z fazy ciekłej do gazowej powoduje przejście innej cząsteczki z fazy gazowej do ciekłej. Proces taki nazywamy kondensacją. Układ znajduje się w stanie równowagi dynamicznej, co oznacza, że mamy do czynienia z taką sama ilością cząsteczek cieczy parujących, jak i ulegających kondensacji . Ilość cząsteczek pary a zatem ciśnienie, nie może już wzrosnąć. O parze znajdującej się w takim stanie mówimy, że jest parą nasyconą. Staje się teraz oczywiste, że w bezwietrzny dzień pomimo świecącego słońca, ubranie schnie dłużej niż w zimniejszy, ale wietrzny dzień. W dzień bezwietrzny przy powierzchni naszego ubrania pojawia się warstwa pary nasyconej i ubranie dalej nie może schnąć (woda w takim samym tempie paruje z ubrania, jak się na nim skrapla). W przypadku wietrznego dnia, podmuchy wiatru porywają pary cieczy i nie tworzy się warstewka pary nasyconej. Woda z ubrania przez cały czas paruje. Szybkość parowania osiąga wartość maksymalną w próżni.

7
Wrzenie Wyobraźmy sobie podobnie jak poprzednio naczynie, którego zawartość jest ogrzewana, czyli, że przez cały czas dostarczamy z zewnątrz energii na sposób ciepła, więcej niż układ jej zużywa na wykonanie pracy objętościowej i pokonanie sił spójności. W związku z tym, średnia prędkość cząsteczek cieczy ciągle wzrasta. Co raz więcej cząsteczek znajduje się w stanie pary, czyli ciśnienie pary jest co raz to większe. Oczywiście ciśnienie pary może maksymalnie wzrosnąć do ciśnienia zewnętrznego. Temperatura w której ciśnienie par zrówna się z ciśnieniem zewnętrznym, nazywana jest temperaturą wrzenia. Podczas wrzenia wewnątrz cieczy, a także na ściankach naczynia powstają pęcherzyki pary nasyconej, zwiększające szybko swoją objętość, które unosząc się ku powierzchni wprawiają ciecz w ruch burzliwy. Cząsteczki te na skutek braku stanu równowagi nie mogą się skroplić i stąd „uciekają" z cieczy. W temperaturze wrzenia, większość cząsteczek cieczy ma odpowiednią prędkość (energię), by pokonać siły spójności. Ale pomimo tego, że cząsteczka z dna naczynia posiada odpowiednią energię, mało prawdopodobnym jest, by pokonała ona drogę od dna naczynia do powierzchni cieczy, nie zderzając się po drodze z inną cząsteczką Dlatego, nawet w temperaturze wrzenia, przejściu w stan pary ulegają jedynie cząsteczki znajdujące się tuż pod powierzchnią cieczy.
, by pokonać siły spójności. Ale pomimo tego, że cząsteczka z dna naczynia posiada odpowiednią energię, mało prawdopodobnym jest, by pokonała ona drogę od dna naczynia do powierzchni cieczy, nie zderzając się po drodze z inną cząsteczką Dlatego, nawet w temperaturze wrzenia, przejściu w stan pary ulegają jedynie cząsteczki znajdujące się tuż pod powierzchnią cieczy..")
8
Wrzenie cieczy następuje gdy ciśnienie par zrówna się z ciśnieniem zewnętrznym. Wynika z tego, że na przykład woda wcale nie musi wrzeć w C (373K). W tej temperaturze ciśnienie par równe jest ciśnieniu zewnętrznemu (1013hPa). Jeżeli ciśnienie zewnętrzne będzie niższe, to ciśnienie par będzie mu równe w niższej temperaturze a co za tym idzie, w niższej temperaturze nastąpi też wrzenie wody. Aby cząsteczka ze stanu ciekłego przeszła w stan pary musi mieć odpowiednio wysoką energię by pokonać siły przyciągające ją przez inne cząsteczki. Nie są to siły przyciągania grawitacyjnego lecz całkiem inne. W stanie ciekłym cząsteczki oddziałują ze sobą na różne sposoby. Im przyciąganie się wzajemne cząsteczek (siły spójności) jest większe, tym trudniej cząsteczkom przejść do fazy gazowej a temperatura wrzenia jest wyższa. By móc oszacować lub porównać temperatury wrzenia różnych związków, należy oszacować wszystkie oddziaływania międzycząsteczkowe i ich wpływ na temperaturę wrzenia.
. W tej temperaturze ciśnienie par równe jest ciśnieniu zewnętrznemu (1013hPa). Jeżeli ciśnienie zewnętrzne będzie niższe, to ciśnienie par będzie mu równe w niższej temperaturze a co za tym idzie, w niższej temperaturze nastąpi też wrzenie wody. Aby cząsteczka ze stanu ciekłego przeszła w stan pary musi mieć odpowiednio wysoką energię by pokonać siły przyciągające ją przez inne cząsteczki. Nie są to siły przyciągania grawitacyjnego lecz całkiem inne. W stanie ciekłym cząsteczki oddziałują ze sobą na różne sposoby. Im przyciąganie się wzajemne cząsteczek (siły spójności) jest większe, tym trudniej cząsteczkom przejść do fazy gazowej a temperatura wrzenia jest wyższa. By móc oszacować lub porównać temperatury wrzenia różnych związków, należy oszacować wszystkie oddziaływania międzycząsteczkowe i ich wpływ na temperaturę wrzenia..")
9
Oddziaływania międzycząsteczkowe
By cząsteczka mogła "wyrwać się" z fazy ciekłej do gazowej musi posiadać odpowiednią energię (prędkość). Pomimo tego, że średnia energia kinetyczna cząsteczki nie zależy od jej masy (E=3/2kT - gdzie k jest stałą Boltzmana k=R/NA) a jedynie od temperatury, to jej prędkość zgodnie ze wzorem na energię kinetyczną E = mv2/2 jest zależna od masy cząsteczki, a to wskazuje na to, że temperatura wrzenia będzie zależała od masy cząsteczkowej. Wpływ masy cząsteczkowej pomimo tego, że ma marginalne znaczenie (o wiele większe znaczenie mają inne czynniki), to wygodnie czasami powoływać się na ten argument, że związek o większej masie molowej ma większą temperaturę wrzenia. Po prostu wraz ze zwiększeniem masy molowej związku, zwiększają się inne oddziaływania od niej zależne, które będą omawiane dalej.
. Pomimo tego, że średnia energia kinetyczna cząsteczki nie zależy od jej masy (E=3/2kT - gdzie k jest stałą Boltzmana k=R/NA) a jedynie od temperatury, to jej prędkość zgodnie ze wzorem na energię kinetyczną E = mv2/2 jest zależna od masy cząsteczki, a to wskazuje na to, że temperatura wrzenia będzie zależała od masy cząsteczkowej. Wpływ masy cząsteczkowej pomimo tego, że ma marginalne znaczenie (o wiele większe znaczenie mają inne czynniki), to wygodnie czasami powoływać się na ten argument, że związek o większej masie molowej ma większą temperaturę wrzenia. Po prostu wraz ze zwiększeniem masy molowej związku, zwiększają się inne oddziaływania od niej zależne, które będą omawiane dalej.")
10
Oddziaływania wynikłe z wielkości cząsteczki
Spróbujmy wykonać następujące doświadczenie. Złączmy dwie płytki szklane, a następnie spróbujmy je rozłączyć. Okazuje się, że do rozłączenia ich potrzebujemy użyć pewnej siły, zaobserwujemy, że płytki jak by się do siebie „kleiły”. Natomiast, jeżeli zamiast płytek szklanych weźmiemy 2 kulki szklane, to oczywiście nie zaobserwujemy zjawiska "klejenia" się kulek. Dzieje się tak dlatego, że powierzchnia styku pomiędzy płytkami jest dużo większa od powierzchni styku pomiędzy kulkami. Podobne zjawisko obserwuje się pomiędzy cząsteczkami. Te, które mają większą powierzchnię będą silniej oddziaływać ze sobą od tych, których kształt jest bardziej kulisty. Zatem związek, którego cząsteczki mają kształt bardziej kulisty będzie miał niższą temperaturę wrzenia od związku którego cząsteczki mają kształt wydłużony. Klasycznym przykładem jest n-pentan i neo-pentan:

11
Podobne zjawisko obserwuje się pomiędzy cząsteczkami
Podobne zjawisko obserwuje się pomiędzy cząsteczkami. Te, które mają większą powierzchnię będą silniej oddziaływać ze sobą od tych, których kształt jest bardziej kulisty. Zatem związek, którego cząsteczki mają kształt bardziej kulisty będzie miał niższą temperaturę wrzenia od związku którego cząsteczki mają kształt wydłużony. Klasycznym przykładem jest n-pentan i neo-pentan: Każde rozgałęzienie łańcucha obniża temperaturę wrzenia.

12
Istnieją przypadki (przy długim łańcuchu), że w związku występują tak silne oddziaływania międzycząsteczkowe, że energia ta jest większa od energii wiązań. Oczywiście związku takiego nie można przeprowadzić w stan pary pod normalnym ciśnieniem, ponieważ wcześniej ulegnie rozkładowi. Wiązania wodorowe Jest to najsilniejsze oddziaływanie międzycząsteczkowe. Jak sama nazwa wskazuje „wiązanie”, że jest ono silne, trudno je rozerwać i dlatego ma największy wpływ na temperaturę wrzenia. Wiązanie wodorowe może tworzyć się jedynie pomiędzy atomem wodoru związanym z silnie elektroujemnym pierwiastkiem (F, O, N) a innym elektroujemnym pierwiastkiem (F, O, N): X – H - - X - H Wiązania wodorowe tworzą takie związki jak: woda H2O , kwasy karboksylowe, ciekły amoniak NH3, białka. Możliwość tworzenia się wiązań wodorowych powoduje zarówno wzrost temperatury wrzenia, jak i rozpuszczalności w wodzie. Związki, które mogą tworzyć wiązania wodorowe zawsze są cieczami o stosunkowo wysokich temperaturach wrzenia.
 a innym elektroujemnym pierwiastkiem (F, O, N): X – H - - X - H. Wiązania wodorowe tworzą takie związki jak: woda H2O , kwasy karboksylowe, ciekły amoniak NH3, białka. Możliwość tworzenia się wiązań wodorowych powoduje zarówno wzrost temperatury wrzenia, jak i rozpuszczalności w wodzie. Związki, które mogą tworzyć wiązania wodorowe zawsze są cieczami o stosunkowo wysokich temperaturach wrzenia.")
13
Para nasycona i nie nasycona
W przypadku kiedy ciecz paruje w stałej temperaturze w zamkniętym, ale nie całkowicie wypełnionym naczyniu, nad powierzchnią cieczy ustala się stan równowagi oznaczający, że szybkość parowania i skraplania się cieczy ulega wyrównaniu. W tym stanie równowagi parowania i skraplania się cieczy występuje zjawisko stałego stężenia par nad powierzchnią cieczy. Para ta nosi nazwę pary nasyconej, a jej ciśnienie określa się jako prężność pary nasyconej. ( Zamknięcie naczynia lub powierzchni, z której paruje ciecz może stanowić otaczająca ją atmosfera.) Ciśnienie pary nasyconej Wartość prężności pary nasyconej w stałej temperaturze jest stała i rośnie wraz ze wzrostem temperatury. W momencie kiedy wartość prężności pary(ciśnienie) zrówna się z ciśnieniem atmosferycznym, charakter parowania cieczy zmienia się zasadniczo, bowiem parowanie nie zachodzi już tylko na powierzchni, ale i w całej objętości cieczy tj. ciecz wrze. Zwiększając ciśnienie zwiększamy temperaturę wrzenia cieczy. Temperatura wrzącej cieczy jest taka sama jak temperatura pary nasyconej , która się z tej cieczy wytwarza. Ciśnienie pary i cieczy jest jednakowe. Istnieje ścisła zależność temperatury pary nasyconej od jej ciśnienia.
 Ciśnienie pary nasyconej. Wartość prężności pary nasyconej w stałej temperaturze jest stała i rośnie wraz ze wzrostem temperatury. W momencie kiedy wartość prężności pary(ciśnienie) zrówna się z ciśnieniem atmosferycznym, charakter parowania cieczy zmienia się zasadniczo, bowiem parowanie nie zachodzi już tylko na powierzchni, ale i w całej objętości cieczy tj. ciecz wrze. Zwiększając ciśnienie zwiększamy temperaturę wrzenia cieczy. Temperatura wrzącej cieczy jest taka sama jak temperatura pary nasyconej , która się z tej cieczy wytwarza. Ciśnienie pary i cieczy jest jednakowe. Istnieje ścisła zależność temperatury pary nasyconej od jej ciśnienia.")
14
Zależność temperatury pary nasyconej od ciśnienia
W zamieszczonej poniżej tabeli w pierwszej kolumnie podane jest ciśnienie, a w drugiej kolumnie temperatura wrzącej wody lub pary nasyconej, odpowiadająca danemu ciśnieniu. Zależność temperatury pary nasyconej od ciśnienia Z danych zawartych w tej tabeli widać, że im ciśnienie w naczyniu jest większe, tym i temperatura wody oraz pary nasyconej jest wyższa. Jeżeli znane jest ciśnienie pary nasyconej w kotle (naczyniu) , zawsze można ustalić, jaka jest temperatura pary i wody, i odwrotnie, znając temperaturę można ustalić wartość ciśnienia.
 , zawsze można ustalić, jaka jest temperatura pary i wody, i odwrotnie, znając temperaturę można ustalić wartość ciśnienia.")
15
Stężenie par nasyconych
Stężenie par nasyconych z punktu widzenia zagrożenia pożarowego jaką stanowi ciecz palna jest podstawą do ustalania jej podstawowych parametrów takich jak: temperatura zapłonu, temperatura zapalenia czy też granic wybuchowości w mieszaninie z powietrzem lub innym utleniaczem. Stężenie par nasyconych cieczy uzależnione jest przede wszystkim od temperatury i ciśnienia. Pojęcie „stężenie par nasyconych” pojawia się w takich definicjach określających parametry cieczy palnych jak: Temperatura zapłonu – jest to najniższa charakterystyczna temperatura dla cieczy palnych, przy której ciecz tworzy nad swoją powierzchnią, mieszaninę par z powietrzem w odpowiednim stosunku, zdolną zapalić się od bodźca energetycznego w określonych warunkach badania. (odpowiedni stosunek – to inaczej sformułowane stężenie par nasyconych) Temperatura zapalenia – jest to najniższa temperatura materiału, który ogrzewany strumieniem ciepła dostarczonym z zewnątrz w wyniku rozkładu termicznego wydziela palną fazę lotną o stężeniu umożliwiającym jego zapalenie się – to znaczy samorzutne pojawienie się płomienia. Dolna granica wybuchowości (DGW) – jest to najniższe stężenie paliwa w mieszaninie palnej, poniżej którego nie jest możliwy jej zapłon pod wpływem czynnika inicjującego ( bodźca energetycznego), w określonych warunkach badania. Górna granica wybuchowości (GGW) – jest to najwyższe stężenie paliwa w mieszaninie palnej, powyżej którego nie jest możliwy jej zapłon pod wpływem czynnika inicjującego (bodźca energetycznego), w określonych warunkach badania.
 Temperatura zapalenia – jest to najniższa temperatura materiału, który ogrzewany strumieniem ciepła dostarczonym z zewnątrz w wyniku rozkładu termicznego wydziela palną fazę lotną o stężeniu umożliwiającym jego zapalenie się – to znaczy samorzutne pojawienie się płomienia. Dolna granica wybuchowości (DGW) – jest to najniższe stężenie paliwa w mieszaninie palnej, poniżej którego nie jest możliwy jej zapłon pod wpływem czynnika inicjującego ( bodźca energetycznego), w określonych warunkach badania. Górna granica wybuchowości (GGW) – jest to najwyższe stężenie paliwa w mieszaninie palnej, powyżej którego nie jest możliwy jej zapłon pod wpływem czynnika inicjującego (bodźca energetycznego), w określonych warunkach badania.")
16
Zależność miedzy ciśnieniem – stężeniem , granicami wybuchowości i temperaturą zapłonu przedstawiono dla etanolu na zamieszczonym poniżej rysunku. Rysunek przedstawia zależność między temperaturą, ciśnieniem pary nasyconej i granicami wybuchowości etanolu. Temperatury odpowiadające dolnej i górnej granicy wybuchowości nazywane są temperaturowymi granicami wybuchowości. Temperatura dolnej granicy jest z reguły równa temperaturze zapłonu. Poniżej tej temperatury ciecze nie stwarzają zagrożenia wybuchowego. Powyżej górnej granicy, pary nasycone nad cieczą tworząc z powietrzem mieszaninę bogatą czyli nie wybuchową.

17
Szybkość parowania Parowanie cieczy – a tym samym szybkość parowania jak już wcześniej wspomniano ma ścisły związek z budową cząsteczki danej cieczy. Szybkość parowania różnych cieczy w jednakowych warunkach jest różna. Podstawowymi czynnikami mającymi wpływ na szybkość parowania, są: 1. temperatura początkowa cieczy 2. średnica zbiornika, 3. poziom cieczy w zbiorniku, 4. stopień zawartości wody w cieczy, 5. wpływ wiatru.

18
Ad.1. Zależność szybkości parowania cieczy, a w konsekwencji szybkości jej spalania od
temperatury początkowej uwarunkowana jest tym, że przy wyższej temperaturze początkowej cieczy ilość ciepła potrzebnego na ogrzanie cieczy do temperatury wrzenia jest mniejsza, a więc ciepło to w znacznej ilości ukierunkowane jest bezpośrednio na parowanie. Ad.2. Zależność szybkości spalania cieczy od średnicy zbiornika wyraża się w fakcie zmniejszania szybkości spalania przy zwiększeniu średnicy zbiornika oraz występowania zjawiska najszybszego spalania się cieczy przy małych średnicach zbiorników ( do około 5m ), przy ściankach tych zbiorników. Ad. 3. Poziom cieczy w zbiorniku ma taki wpływ na szybkość spalania cieczy, że wraz z obniżeniem się poziomu cieczy, szybkość jej spalania nieznacznie maleje. Ad. 4. Podobne zjawisko zachodzi przy nawilgoceniu cieczy, co tłumaczy się tym, że podczas spalania, znaczna ilość ciepła skierowana jest na wyparowanie wody. Ad. 5. Wpływ wiatru powoduje zwiększenie się szybkości spalania cieczy, głównie na skutek zasilania płomieni dużą ilością tlenu i przyciskania płomienia do powierzchni cieczy, przez co otrzymuje ona znacznie większą ilość ciepła niż przy pogodzie bezwietrznej i tak np. przy wzroście szybkości wiatru od 0 do 8 m/s, szybkość spalania benzyny w zbiorniku wzrasta prawie o 50%.
, przy ściankach tych zbiorników. Ad. 3. Poziom cieczy w zbiorniku ma taki wpływ na szybkość spalania cieczy, że. wraz z obniżeniem się poziomu cieczy, szybkość jej spalania nieznacznie maleje. Ad. 4. Podobne zjawisko zachodzi przy nawilgoceniu cieczy, co tłumaczy się tym, że. podczas spalania, znaczna ilość ciepła skierowana jest na wyparowanie wody. Ad. 5. Wpływ wiatru powoduje zwiększenie się szybkości spalania cieczy, głównie na. skutek zasilania płomieni dużą ilością tlenu i przyciskania płomienia do. powierzchni cieczy, przez co otrzymuje ona znacznie większą ilość ciepła niż. przy pogodzie bezwietrznej i tak np. przy wzroście szybkości wiatru. od 0 do 8 m/s, szybkość spalania benzyny w zbiorniku wzrasta prawie o 50%.")
19
Temperatura zapłonu cieczy jednorodnych i niejednorodnych
Temperatura zapłonu – jest to najniższa charakterystyczna temperatura dla cieczy palnych, przy której ciecz tworzy nad swoją powierzchnią, mieszaninę par z powietrzem w odpowiednim stosunku, zdolną zapalić się od bodźca energetycznego w określonych warunkach badania. Temperatura zapłonu została uznana jako kryterium oceny substancji palnych z punktu widzenia zagrożenia pożarowego i jest podstawą ich klasyfikacji. Jest ona również wielkością charakterystyczną dla ciał stałych jednolitej strukturze chemicznej, jak np. dla naftalenu, fosforu lub kamfory, które wytwarzają pary (sublimują) w ilości wystarczającej do zapłonu poniżej ich temperatury topnienia. Temperatura zapłonu jest parametrem, dla którego zostały opracowane znormalizowane warunki pomiaru. Przez temperaturę zapłonu należy rozumieć najniższą temperaturę, przy której ogrzana ciecz tworzy nad swą powierzchnią, w odpowiednim stosunku jej par z powietrzem mieszaninę palną, zdolną zapalić się na krótką chwilę od płomyka przesuniętego tuż nad powierzchnią cieczy. Zapalenie to charakteryzuje się lekkim wybuchem (fuknięciem), po czym płomień gaśnie szybko i nie rozprzestrzenia się po zbliżeniu bodźca termicznego. Jak wynika z definicji, ten parametr określający stopień niebezpieczeństwa pożarowego cieczy oparty jest na mechanizmie przejścia cieczy w parę oraz łączenia się tej cieczy z powietrzem. Od tej zasady istnieją też wyjątki, z których przykładowo można wskazać na eter etylowy i biosiarkowy, które ze względu na tworzenie przy dłuższym przechowywaniu nadtlenków mogą być wybuchowe i zapalne bez konieczności uprzedniego przejścia w stan pary.
 w ilości wystarczającej do zapłonu poniżej ich temperatury topnienia. Temperatura zapłonu jest parametrem, dla którego zostały opracowane znormalizowane warunki pomiaru. Przez temperaturę zapłonu należy rozumieć najniższą temperaturę, przy której ogrzana ciecz tworzy nad swą powierzchnią, w odpowiednim stosunku jej par z powietrzem mieszaninę palną, zdolną zapalić się na krótką chwilę od płomyka przesuniętego tuż nad powierzchnią cieczy. Zapalenie to charakteryzuje się lekkim wybuchem (fuknięciem), po czym płomień gaśnie szybko i nie rozprzestrzenia się po zbliżeniu bodźca termicznego. Jak wynika z definicji, ten parametr określający stopień niebezpieczeństwa pożarowego cieczy oparty jest na mechanizmie przejścia cieczy w parę oraz łączenia się tej cieczy z powietrzem. Od tej zasady istnieją też wyjątki, z których przykładowo można wskazać na eter etylowy i biosiarkowy, które ze względu na tworzenie przy dłuższym przechowywaniu nadtlenków mogą być wybuchowe i zapalne bez konieczności uprzedniego przejścia w stan pary.")
20
Należy podkreślić, że temperatura zapłonu określana jest zgodnie z PN przy ciśnieniu
760 mmHg, W związku z tym jest oczywiste , że przy innych wartościach ciśnienia zewnętrznego, temperatura faktyczna zapłonu cieczy może wzrastać lub maleć. Jest to bardzo istotne w toku analizy procesów technologicznych, gdzie ciśnienie zewnętrzne może mieć inną wartość i w której to sytuacji należy ustalić laboratoryjnie , w jakim stopniu faktyczne ciśnienie w danym miejscu może mieć wpływ na kształtowanie się faktycznej temperatury zapłonu par cieczy palnych. Podkreślić tu trzeba, że zdolność do zapłonu mają tylko mieszaniny palne o określonym składzie. Zakres stosunków paliwa do utleniacza, w którym występuje zapłon mieszanki jest określony dwiema granicami, ubogą - o niedomiarze paliwa i bogatą - o jego nadmiarze. Na przykład mieszanina dwutlenku węgla z powietrzem (przy ciśnieniu atmosferycznym) jest zdolna do zapłonu i palenia się w granicach od 12,5 do 74,2 % udziału masowego CO w powietrzu. Istnienie granic zapłonu tłumaczy cieplna teoria zapłonu: na granicy ubogiej mieszaniny ma duży balast w postaci utleniacza, na granicy bogatej natomiast - w postaci paliwa. W rezultacie ilość ciepła wydzielona w procesie reakcji nie jest w stanie przewyższyć ilości ciepła zużytkowanego na ogrzanie czynnika nie biorącego udziału w reakcji i proces spalania nie może rozprzestrzenić się samoczynnie. Na granice zapłonu ma wpływ wiele czynników: początkowe ciśnienie i temperatura mieszanki, energia źródeł, udział domieszek itp. Wraz ze zmniejszeniem się ciśnienia granice zapłonu zwężają się aż do punktu, poniżej którego zapłon jest już niemożliwy. Wzrost początkowej temperatury mieszaniny palnej rozszerza granice zapłonu. Oddziaływanie dodatków może powodować rozszerzenie bądź zwężenie tych granic, a zwłaszcza granicy bogatej, co tłumaczy się zmniejszeniem udziału tlenu w mieszaninie.
 jest zdolna do zapłonu i palenia się w granicach od 12,5 do 74,2 % udziału masowego CO w powietrzu. Istnienie granic zapłonu tłumaczy cieplna teoria zapłonu: na granicy ubogiej mieszaniny ma duży balast w postaci utleniacza, na granicy bogatej natomiast - w postaci paliwa. W rezultacie ilość ciepła wydzielona w procesie reakcji nie jest w stanie przewyższyć ilości ciepła zużytkowanego na ogrzanie czynnika nie biorącego udziału w reakcji i proces spalania nie może rozprzestrzenić się samoczynnie. Na granice zapłonu ma wpływ wiele czynników: początkowe ciśnienie i temperatura mieszanki, energia źródeł, udział domieszek itp. Wraz ze zmniejszeniem się ciśnienia granice zapłonu zwężają się aż do punktu, poniżej którego zapłon jest już niemożliwy. Wzrost początkowej temperatury mieszaniny palnej rozszerza granice zapłonu. Oddziaływanie dodatków może powodować rozszerzenie bądź zwężenie tych granic, a zwłaszcza granicy bogatej, co tłumaczy się zmniejszeniem udziału tlenu w mieszaninie.")
21
Granice zapłonu mieszanin, w których skład wchodzi wiele paliw można określić z zależności: /L = n1/N1 + n2/N2 + n3/N Gdzie: L granica zapłonu uboga lub bogata (wyrażona w % udziału objętościowego gazu palnego złożonego w mieszaninie), n1, n2, n3 - udział poszczególnych gazów palnych w całej masie paliwa, N1, N2, N3 - granica zapłonu składników palnych mieszaniny (uboga lub bogata). Granice zapłonu niektórych mieszanin palnych
, n1, n2, n3 - udział poszczególnych gazów palnych w całej masie paliwa, N1, N2, N3 - granica zapłonu składników palnych mieszaniny. (uboga lub bogata). Granice zapłonu niektórych mieszanin palnych.")
22
na skutek mniejszej powierzchni strat cieplnych,
Podobnie, jak w przypadku temperatury zapalenia , również i na temperaturę zapłonu mają wpływ omówione wcześniej parametry, jak wilgotność, ciśnienie itp., przy czym dla cieczy bardzo poważny wpływ na kształtowanie się temperatury zapłonu ma także średnica i objętość naczynia zawierającego ciecz oraz właściwości katalityczne ścianek tych naczyń, gdzie zależności są następujące: 1. im mniejsza jest objętość naczynia, tym niższa jest temperatura zapłonu, a to na skutek mniejszej powierzchni strat cieplnych, 2. katalityczne własności ścianek naczynia mogą być zarówno dodatnie jak i ujemne, tzn. przyspieszające reakcję utleniania, a więc zmniejszające temperaturę zapłonu lub opóźniające tę reakcję, tj. podnoszące temperaturę zapłonu.

23
Klasyfikacja cieczy palnych
Temperatura zapłonu cieczy jest najważniejszym tzw. parametrem pożarowym dla cieczy. Jest ona podstawą klasyfikacji cieczy na trzy klasy niebezpieczeństwa pożarowego. Klasa I ciecze o temperaturze zapłonu do 230C. Klasa II ciecze o temperaturze zapłonu >230C do 61 0C. Klasa III ciecze o temperaturze zapłonu > 61 0C do 100 0C Ciecze mające temperaturę zapłonu do 61 0C (I i II klasa) klasyfikowane są jako ciecze niebezpieczne pożarowo. Pod pojęciem cieczy palnej rozumnie się ciecz o temperaturze zapłonu od 610C do 1000C. Jest to oczywiście klasyfikacja umowna. Temperatury zapalenia cieczy mieszczą się na ogól w granicach C .Wyjątki to: eter C, dwusiarczek węgla 102 0C, benzyna lotnicza 2600C i inne.
 klasyfikowane są jako ciecze niebezpieczne pożarowo. Pod pojęciem cieczy palnej rozumnie się ciecz o temperaturze zapłonu od 610C do 1000C. Jest to oczywiście klasyfikacja umowna. Temperatury zapalenia cieczy mieszczą się na ogól w granicach C .Wyjątki to: eter 160 0C, dwusiarczek węgla 102 0C, benzyna lotnicza 2600C i inne.")
24
W tabeli podano przykłady temperatur zapłonu i zapalenia różnych cieczy.

25
Mechanizm zapalenia i palenia cieczy palnych
Spalanie cieczy zachodzi wtedy, gdy nad powierzchnią cieczy znajduje się zdolna do spalania mieszanina par cieczy z powietrzem. Jeżeli stężenie par jest niższe od stężenia odpowiadającego dolnej granicy zapalności lub jeśli jest za mało powietrza np. rozrzedzenie par gazem obojętnym) spalanie cieczy jest praktycznie niemożliwe. Aby powstała palna mieszanina par cieczy z powietrzem zapaliła się, ciecz musi mieć temperaturę nie niższą niż jej temperatura zapłonu, a ilość tlenu w mieszaninie nie może być mniejsza niż 12% obj. Spalanie mieszaniny może nastąpić w obecności źródła ciepła posiadającego odpowiednią energię cieplną. Warunki konieczne do zapalenia się cieczy w ujęciu schematycznym
 spalanie cieczy jest praktycznie niemożliwe. Aby powstała palna mieszanina par cieczy z powietrzem zapaliła się, ciecz musi mieć temperaturę nie niższą niż jej temperatura zapłonu, a ilość tlenu w mieszaninie nie może być mniejsza niż 12% obj. Spalanie mieszaniny może nastąpić w obecności źródła ciepła posiadającego odpowiednią energię cieplną. Warunki konieczne do zapalenia się cieczy w ujęciu schematycznym.")
26
Warunki konieczne dla zaistnienia spalania cieczy przedstawiono na rysunku. Kiedy temperatura cieczy jest równa lub wycisza od jej temperatury zapłonu, mieszanina par z powietrzem istniejąca nad cieczą ulega zapłonowi przy wprowadzeniu do tej mieszaniny źródła ciepła, posiadającego niezbędny zapas ciepła do ogrzania mieszaniny do tej temperatury. Po zapaleniu, gdy temperatura górnej warstwy cieczy jest wyższa od temperatury zapłonu, ustala się spalanie ciągle. Proces ciągłego palenia cieczy w ujęciu schematycznym

27
Podczas spalania cieczy powstaje płomień, w którym ma miejsce energetyczne przygotowanie pary cieczy do spalania (ogrzania, dysocjacja termiczna) a następnie mieszanie otrzymanych produktów z powietrzem, utlenianie i spalanie. Spalanie mieszaniny przebiega w cienkiej warstwie strefy spalania. Do strefy spalania z powierzchni cieczy dyfundują pary, a z powietrza do płomienia dyfunduje tlen. Temperatura strefy spalania w każdym punkcie strefy jest wyższa od temperatury zapalenia mieszaniny, dlatego strefa spalania jest stałym źródłem zapalenia. Konieczne do parowania ciepło jest przekazywane drogą promieniowania od strefy spalania płomienia do powierzchni cieczy. W ten sposób w procesie spalania cieczy ustala się równowaga pomiędzy ilością ciepła przekazywaną drogą promieniowania ze strefy spalania do powierzchni cieczy, a ilością ciepła zużywaną na powstanie par dyfundujących z powierzchni cieczy do płomienia. Jeśli ta równowaga zostanie naruszona, proces spalania cieczy zostaje przerwany.

28
Mechanizm spalania cieczy
Przekazywanie ciepła do ze strefy palenia do powierzchni cieczy następuje głównie drogą promieniowania. Przekazywanie ciepła drogą przewodnictwa cieplnego par ze względu na ich dużą szybkość ruchu od powierzchni cieczy do strefy spalania nie jest możliwe. Przekazywanie ciepła drogą konwekcji także nie jest możliwe, ponieważ strumień par w objętości płomienia jest ukierunkowany od powierzchni mniej nagrzanej (cieczy) do warstwy gazów bardziej nagrzanej (strefy palenia. Ilość ciepła przekazywana od strefy palenia się nie jest stała. Zależy ona od dokładności spalania, jaskrawości płomienia, odległości pomiędzy strefą palenia a powierzchnią cieczy, kształtu płomienia itp. Na całkowite spalanie cieczy bardzo duży wpływ wywiera wiatr. Z przytoczonych w tej tabeli danych widać, że przy zmianie prędkości wiatru od 0 do 2 m/s temperatura płomienia podwyższa się o 700 C. Powoduje to zwiększenie intensywności świecenia płomienia, a co za tym idzie – zwiększenie ilości ciepła przekazywanego drogą promieniowania do powierzchni palącej się cieczy. Prędkość wiatru w m/s Cisza 08 1,0 2,0 Temperatura płomienia w 0 C 1120 1180 1190 Zmiany temperatury płomienia benzyny w zależności od prędkości wiatru
 do warstwy gazów bardziej nagrzanej (strefy palenia. Ilość ciepła przekazywana od strefy palenia się nie jest stała. Zależy ona od dokładności spalania, jaskrawości płomienia, odległości pomiędzy strefą palenia a powierzchnią cieczy, kształtu płomienia itp. Na całkowite spalanie cieczy bardzo duży wpływ wywiera wiatr. Z przytoczonych w tej tabeli danych widać, że przy zmianie prędkości wiatru od. 0 do 2 m/s temperatura płomienia podwyższa się o 700 C. Powoduje to zwiększenie intensywności świecenia płomienia, a co za tym idzie – zwiększenie ilości ciepła przekazywanego drogą promieniowania do powierzchni palącej się cieczy. Prędkość wiatru w m/s. Cisza ,0. 2,0. Temperatura płomienia w 0 C Zmiany temperatury płomienia benzyny w zależności od prędkości wiatru.")
29
Poza ciepłem otrzymywanym drogą promieniowania ciecz nagrzewana jest również przez ścianki zbiornika. Nagrzewanie to jest najbardziej intensywne wówczas , gdy poziom cieczy jest niższy od górnej krawędzi zbiornika. Ciepło dochodzące do powierzchni cieczy zużywa się na ogrzanie jej i parowanie. Parowanie cieczy podczas palenia niczym się nie różni od jej zwykłego parowania, jest tylko bardziej intensywne. Jeżeli pali się ciecz jednorodna, to skład pary nie różni się od składu cieczy. Jeżeli natomiast pali się ciecz niejednorodna , to skład pary nie jest jednakowy ze składem cieczy. Do grupy cieczy niejednorodnych zalicza się naftę i wszystkie produkty naftowe. Podczas ich palenia w większym stopniu następuje parowanie frakcji lekkich, w rezultacie czego ciecz zmienia swój skład i właściwości. Podczas palenia się mieszaniny cieczy palnych z wodą, w rezultacie parowania zwiększa się w tej cieczy ilość wody. Woda bowiem paruje wolniej niż większość cieczy palnych, co prowadzi do wzrostu ciężaru właściwego mieszaniny cieczy palnej. Proces ten jest charakterystyczny dla mieszanin, w których składnik palny ma temperaturę wrzenia niższą od temperatury wrzenia wody. Przy długotrwałym spalaniu mieszaniny cieczy palnych z wodą procent wody zwiększa się i następuje moment, kiedy palenie przerywa się, chociaż nie cała mieszanina się wypaliła. Mieszanina cieczy palnych z wodą, jeżeli temperatura wrzenia cieczy palnej jest wyższa od temperatury wrzenia wody, zachowuje się podczas palenia w ten sposób, że w miarę jej spalania zmniejsza się w niej procent zawartości wody. Taka mieszanina wypala się całkowicie.

30
Szybkość spalania - szybkość wagowa i liniowa
Liniowa szybkość spalania – określa o ile obniży się poziom cieczy w zbiorniku w określonym czasie [mm/min] Wagowa szybkość spalania – określa ubytek masy cieczy z powierzchni w jednostce czasu [ kg/m2xgodz ]. W zamieszczonej poniżej tabeli podano szybkości spalania i tzw. Obciążenie cieplne powierzchni parowania cieczy to znaczy ilość ciepła [kJ] przekazywaną z płomienia na jednostkę powierzchni spalania [m2] w jednostce czasu [godz] Na podstawie badań stwierdzono, że: 1. Niezależnie od sposobu ogrzewania cieczy, ilość ciepła, wypromieniowana przez płomień dyfuzyjny do powierzchni palącej się cieczy (tzw. obciążenie cieplne), jest wielkością stalą, charakterystyczną dla danej cieczy, 2. Podczas spalania się cieczy obciążenie cieplne powierzchni parowania zależy tylko od właściwości fizyczno-chemicznych cieczy (przy jednakowych warunkach zewnętrznych), 3. Największe obciążenie cieplne powierzchni parowania mają te ciecze, które podczas spalania dają płomień silnie kopcący, charakteryzujący się dużą zdolnością promieniowania ciepła, 4. Szybkość spalania cieczy rośnie wraz ze wzrostem temperatury i wzrostem obciążenia cieplnego, a zmniejsza się wraz ze wzrostem ciepła parowania cieczy i ciepła właściwego,
, jest wielkością stalą, charakterystyczną. dla danej cieczy, 2. Podczas spalania się cieczy obciążenie cieplne powierzchni parowania. zależy tylko od właściwości fizyczno-chemicznych cieczy. (przy jednakowych warunkach zewnętrznych), 3. Największe obciążenie cieplne powierzchni parowania mają te ciecze, które podczas spalania dają płomień silnie kopcący, charakteryzujący. się dużą zdolnością promieniowania ciepła, 4. Szybkość spalania cieczy rośnie wraz ze wzrostem temperatury. i wzrostem obciążenia cieplnego, a zmniejsza się wraz ze wzrostem. ciepła parowania cieczy i ciepła właściwego,")
31
5. Szybkość spalania się cieczy osiąga wartość maksymalną, jeśli wstępnie, przed spalaniem, ciecz ogrzana jest do temperatury wrzenia, a minimalną, jeśli przed spaleniem ciecz ma temperaturę równą temperaturze krzepnięcia. Szybkość spalania i obciążenie cieplne powierzchni parowania cieczy

32
Liniowa szybkość spalania – określa o ile obniży się poziom cieczy w zbiorniku w określonym czasie [mm/min] i określana jest wzorem: VLc – liniowa szybkość wypalania cieczy w mm/min H - grubość spalonej warstwy w mm tpal - czas trwania palenia w min. Liniową szybkość wypalania można także określić z szybkości wagowej, o ile znana jest masa właściwa palącej się cieczy przy określonej temperaturze spalania: Vm – szybkość spalania masy wagowej w kg/m2 x min v – masa właściwa palącej się cieczy przy określonej temperaturze palenia w kg/m3. Znając powierzchnie zbiornika (powierzchnię lustra cieczy) i grubość spalonej warstwy możemy określić ubytek jednostki objętości cieczy w czasie [ml/min] mnożąc powierzchnię lustra cieczy przez grubość spalonej warstwy.
![Liniowa szybkość spalania – określa o ile obniży się poziom cieczy w zbiorniku w określonym czasie [mm/min] i określana jest wzorem:](http://slideplayer.pl/slide/10184559/33/images/32/Liniowa+szybko%C5%9B%C4%87+spalania+%E2%80%93+okre%C5%9Bla+o+ile+obni%C5%BCy+si%C4%99+poziom+cieczy+w+zbiorniku+w+okre%C5%9Blonym+czasie+%5Bmm%2Fmin%5D+i+okre%C5%9Blana+jest+wzorem%3A.jpg "VLc – liniowa szybkość wypalania cieczy w mm/min. H - grubość spalonej warstwy w mm tpal - czas trwania palenia w min. Liniową szybkość wypalania można także określić z szybkości wagowej, o ile znana jest masa właściwa palącej się cieczy przy określonej temperaturze spalania: Vm – szybkość spalania masy wagowej w kg/m2 x min. v – masa właściwa palącej się cieczy przy określonej temperaturze palenia w kg/m3. Znając powierzchnie zbiornika (powierzchnię lustra cieczy) i grubość spalonej warstwy możemy określić ubytek jednostki objętości cieczy w czasie [ml/min] mnożąc powierzchnię lustra cieczy przez grubość spalonej warstwy.")
33
Przegrzewanie się cieczy palnych w procesie palenia
Podczas pożarów najczęściej ma miejsce spalanie cieczy lub mieszanin cieczy ze swobodnej powierzchni. Spalanie cieczy ze swobodnej powierzchni charakteryzuje się tym, że oprócz ogrzewania górnej warstwy cieczy obserwuje się przegrzewanie głębiej położonych warstw paliwa ciekłego. W czasie spalania cieczy jednorodnych np. alkoholi, acetonu, benzenu i innych niskocząsteczkowych cieczy stała szybkość spalania ustala się bardzo szybko po rozpoczęciu spalania. Górna warstwa cieczy ogrzewa się do temperatury wrzenia, i nie obserwuje się znacznego przegrzewania się warstwy cieczy, położonej głębiej w zbiorniku. W czasie spalania w takich warunkach, niewielka warstewka przegrzana znajdująca się tuż pod powierzchnią nie ulega powiększeniu i ma stałą grubość. Wcześniej już zaznaczono, że ciepło dochodzące z płomienia do powierzchni cieczy jest zużywane na ogrzanie powierzchni cieczy do jej temperatury wrzenia i odparowanie cieczy. Jeśli szybkość parowania cieczy jest niewielka, to ciepło otrzymywane z płomienia jest zużywane przede wszystkim na ogrzanie cieczy Co powoduje w konsekwencji jej przegrzewanie w głąb. W czasie spalania szybkość parowania cieczy jest równoznaczna z szybkością spalania ze swobodnej powierzchni. W związku z powyższym przegrzewanie się cieczy w głąb jest tym większe im mniejsza szybkość spalania. Wszystkie łatwo zapalne ciecze jednorodne mają znacznie większą szybkość spalania niż szybkość przegrzewania, dlatego podczas ich spalania obserwuje się nieznaczną stalą szybkość przegrzewania w głąb.

34
Przykład cieczy (butanol), która podczas spalania nie tworzy warstwy przegrzanej
Ciecze niejednorodne o dużej lepkości (mazut, ropa naftowa itp.) mają niewielką szybkość spalania ze swobodnej powierzchni w porównaniu z szybkością przegrzewania (tabela) dlatego też tworząca się warstwa przegrzewana w czasie trwania spalania powiększa się i przesuwa w głąb.
 mają niewielką szybkość spalania ze swobodnej powierzchni w porównaniu z szybkością przegrzewania (tabela) dlatego też tworząca się warstwa przegrzewana w czasie trwania spalania powiększa się i przesuwa w głąb.")
35
Trzy etapy tworzenia się i rozprzestrzeniania gorącej warstwy
podczas pożaru oleju napędowego w zbiorniku ( t1 < t2 < t3 )
")
36
Spalanie się cieczy niejednorodnych o dużej lepkości
Praktycznie wszystkie typowe paliwa spalają się w ten sposób, że w trakcie pożaru grubość warstwy przegrzanej wzrasta, zwiększając możliwość zaistnienia wielu zjawisk m.in. wykipienia cieczy i jej wyrzutu ze zbiornika. Wskutek takiego przebiegu zjawiska w warunkach pożarowych, ze zbiorników np. produktów naftowych może nastąpić zjawisko wykipienia cieczy (stopniowego zwiększania objętości cieczy i przelewania się jej przez krawędzie zbiornika) lub wyrzutu cieczy na zewnątrz zbiornika.
 lub wyrzutu cieczy na zewnątrz zbiornika.")
37
Zjawisko kipienia jest prawdopodobne w przypadku cieczy o dużej lepkości, zawierającej zemulgowane nierozpuszczalne substancje o niskiej temperaturze wrzenia. Najczęściej substancją tą jest woda, której obecność w produktach naftopochodnych jest związana z procesem wydobywania ropy naftowej. Zawartość wody w ropie w zależności od jej pochodzenia i wydobycia, waha się od 0,03% do około 1%. W czasie magazynowania i w początkowym okresie spalania woda jest mniej lub bardziej równomiernie rozłożona, w masie cieczy. W wyniku zmniejszenia lepkość górnej warstwy cieczy na skutek nagrzewania się, zawieszone krople wody stopniowo opadają, zatrzymując się w głębszych warstwach cieczy o stosunkowo dużej lepkości. Jednocześnie krople wody nagrzewają się i gdy osiągną określoną temperaturę wówczas wyparowują. Wytwarzająca się para wodna powoduje spienienie cieczy, która przelewa się przez krawędź zbiornika. Jeśli kropelki wody utworzą na dnie zbiornika z cieczą palną warstwę wody, wtedy po osiągnięciu przez warstwę przegrzaną temperatury ponad 100 0C, woda przemienia się szybko w parę. Wskutek ogromnego wzrostu objętości powstałej pary w stosunku do objętości wody, z której ona powstała, następuje wyrzucanie zawartości cieczy palnej istniejącej w zbiorniku i w konsekwencji rozprzestrzenienie się pożaru na znacznie większą powierzchnię. Najczęściej kipi ropa naftowa i mazut, a więc produkty o stosunkowo dużej lepkości umożliwiającej zatrzymanie zawieszonych w nich kropel wody. Stwierdzono, że zależnie od spalającego się produktu istnieje dolna granica wilgotności, poniżej której wykipienie i wyrzut nie jest możliwy (poniżej 0,1% wody). Gdy zawartość wody jest większa niż 20% powstająca emulsja nie pali się.
. Gdy zawartość wody jest większa niż 20% powstająca emulsja nie pali się..")
38
Wrzenie i wyrzut ze zbiorników podczas palenia
Ciecze w zbiornikach nagrzewające się głęboko przez konwekcję burzliwą mają tendencję do wykipienia. Sam mechanizm wykipienia przedstawia się następująco : wzrost temperatury cieczy osłabia jej lepkość, skutkiem czego zawarta w niej woda opada do warstw dolnych i po nagrzaniu się do temperatury powyżej 100 °C paruje (1 litr wody =1700 l pary wodnej). Para wodna unosząc się ku górze porywa ze sobą krople cieczy tworząc rodzaj piany emulsyjnej. Gdy piana ta wydostaje się na powierzchnię lustra cieczy zapala się i pieniąc przelewa się przez brzegi zbiornika. Podczas wykipienia cieczy znacznie pogarsza się sytuacja pożarową, ponieważ stwarza to niebezpieczeństwo dla sił i środków uczestniczących w akcji oraz zagraża sąsiednim zbiornikom. Wykipienie cieczy palnych następuje przeważnie po 45 min. od chwili zapalenia.
. Para wodna unosząc się ku górze porywa ze sobą krople cieczy tworząc rodzaj piany emulsyjnej. Gdy piana ta wydostaje się na powierzchnię lustra cieczy zapala się i pieniąc przelewa się przez brzegi zbiornika. Podczas wykipienia cieczy znacznie pogarsza się sytuacja pożarową, ponieważ stwarza to niebezpieczeństwo dla sił i środków uczestniczących w akcji oraz zagraża sąsiednim zbiornikom. Wykipienie cieczy palnych następuje przeważnie po 45 min. od chwili zapalenia.")
39
Wykipienie palącej się cieczy
Rozprzestrzenianie się pożaru po wykipieniu cieczy

40
Wyrzut cieczy ze zbiornika
Podczas pożarów zbiorników możemy również spotkać się z wyrzutem cieczy ze zbiornika. Sam wyrzut cieczy uwarunkowany jest następującymi czynnikami: 1. obecnością „poduszki wodnej", 2. wysokim wskaźnikiem lepkości cieczy, 3. nagrzewaniem się niższych warstw cieczy do temperatury powyżej 100 °C( na styku z poduszką wodną ). Warunki takie istnieją przede wszystkim w zbiornikach ropy naftowej. Proces nagrzewania się głębokich warstw cieczy jest podobny do tego, który powoduje wykipienie cieczy. Różnica polega na tym, że wysoka lepkość cieczy nie pozwala na unoszenie się pary wodnej. Nad poduszką wodną tworzy się pęcherz par, który pod wpływem ciśnienia przebija się w końcu ku górze porywając ze sobą krople cieczy. Wyrzut cieczy ze zbiornika może mieć zasięg nawet do 200m. W momencie pierwszego wyrzutu ciecz unosi się nad zbiornikiem do wysokości m. Dalsze wyrzuty o rosnącej sile następują w krótkich odstępach czasu. Na czas wyrzutu wpływ ma rodzaj cieczy oraz kształt i średnica zbiornika. W zbiornikach o dużych średnicach, wyrzuty cieczy występują później niż w zbiornikach o średnicach mniejszych. Wyrzuty cieczy nie występują raczej przed upływem 2 godzin od chwili powstania pożaru.
. Warunki takie istnieją przede wszystkim w zbiornikach ropy naftowej. Proces nagrzewania się głębokich warstw cieczy jest podobny do tego, który powoduje wykipienie cieczy. Różnica polega na tym, że wysoka lepkość cieczy nie pozwala na unoszenie się pary wodnej. Nad poduszką wodną tworzy się pęcherz par, który pod wpływem ciśnienia przebija się w końcu ku górze porywając ze sobą krople cieczy. Wyrzut cieczy ze zbiornika może mieć zasięg nawet do 200m. W momencie pierwszego wyrzutu ciecz unosi się nad zbiornikiem do wysokości m. Dalsze wyrzuty o rosnącej sile następują w krótkich odstępach czasu. Na czas wyrzutu wpływ ma rodzaj cieczy oraz kształt i średnica zbiornika. W zbiornikach o dużych średnicach, wyrzuty cieczy występują później niż w zbiornikach o średnicach mniejszych. Wyrzuty cieczy nie występują raczej przed upływem 2 godzin od chwili powstania pożaru.")
41
Postępowanie podczas gaszenia pożarów zbiorników cieczy mogących doprowadzi do wykipienia lub wyrzutu : należy ustalić rodzaj płonącej cieczy ( możliwość jej wykipienia lub wyrzutu), orientacja w możliwych kierunkach rozpływania się przelanej cieczy, stała obserwacja płonącego zbiornika, określić należy kierunki i drogi ewentualnej ucieczki oraz sygnały alarmowe, pracującym przy ochładzaniu, nie wolno kierować wody do wnętrza płonącego zbiornika, wozy bojowe i pompy nie powinny znajdować się bliżej niż 100 m od płonącego zbiornika od strony nawietrznej i 150 m od strony zawietrznej. Płonące zbiorniki zaczynamy gasić, gdy na miejscu mamy już wystarczającą ilość sił i środków.

42
Pożary cieczy w zbiornikach otwartych oraz uwolnionych ze zbiorników zamkniętych
Pożary cieczy i gazów stanowią duże zagrożenie w przemyśle chemicznym, szczególnie petrochemicznym. Nieszczelności i uszkodzenia zbiorników mogą powodować wycieki. Trudne do opanowania są pożary płynących rozlewisk o dużej powierzchni a także pożary cieczy spływającej po powierzchniach pionowych. Wyciek pionowy po zbiorniku znacznie intensyfikuje szybkość spalania. Szybkość propagacji płomienia wzrasta na powierzchniach pionowych do kilkudziesięciu razy w stosunku do szybkości uzyskiwanych na powierzchniach poziomych. Pożary cieczy w zbiornikach stanowią bardzo poważne zagrożenie i stwarzają duże trudności w gaszeniu. Ogrzanie cieczy w zbiorniku zamkniętym, np. w środowisku pożaru może spowodować, że wzrost ciśnienia w zbiorniku i gromadzące się pary oraz wrząca ciecz zostaną gwałtownie wyrzucone na zewnątrz i powstanie tzw. kula ognista (BLEVE)
")
43
Typy pożarów cieczy i gazów palnych
Zdarzenie Typ Charakter spalania Główna przyczyna Pożar Powierzchniowy Spalanie palnej substancji ze swobodnej powierzchni Wyciek z instalacji lub na uszczelnieniu pomp Strumieniowy Zapłon gazu wypływającego ze zbiornika ciśnieniowego przez mały otwór(1-5cm2) Jak wyżej przy nadciśnieniu w zbiorniku Błyskawiczny Deflagracja spalania mieszaniny gaz-powietrze bez wytworzenia niszczącej fali ciśnienia Wyciek cieczy przegrzanej Kulisty Spalanie obłoków paliwo-powietrze tworząc kulistą przestrzeń płomienia Pęknięcie zbiornika z gazem skroplonym wskutek wewnętrznego pożaru typu powierzchniowego lub strumieniowego
 Jak wyżej przy nadciśnieniu w zbiorniku. Błyskawiczny. Deflagracja spalania mieszaniny gaz-powietrze bez wytworzenia niszczącej fali ciśnienia. Wyciek cieczy przegrzanej. Kulisty. Spalanie obłoków paliwo-powietrze tworząc kulistą przestrzeń płomienia. Pęknięcie zbiornika z gazem skroplonym wskutek wewnętrznego pożaru typu powierzchniowego lub strumieniowego.")
44
Pożar powierzchniowy (spalanie cieczy ze swobodnej powierzchni)
Spalanie cieczy ma charakter dyfuzyjny i przebiega w układzie pary palne-powietrze. Dynamika spalania cieczy w zbiornikach jest zależna od: lotności cieczy, średnicy zbiornika, przebiegu nagrzewania się cieczy w głąb warstwy, zawartości w cieczy wody emulgowanej lub odstanej na dnie, warunków atmosferycznych, szczególnie siły wiatru.

45
Pożar strumieniowy Powstaje, gdy uwalniająca się ze zbiornika lub rurociągu ciśnieniowego ciecz palna lub gaz ulegnie zapłonowi. Powstaje wtedy długi stabilny płomień, przypominający płomień z palników do cięcia lub spawania metali. Strumień cieplny emitowany przez taki płomień jest nadzwyczaj intensywny i powoduje duże obciążenia cieplne elementów konstrukcyjnych i sprzętu. Zależy ono od długości płomienia, który jest funkcją prędkości wypływu oraz odległości elementu od miejsca wypływu.

46
Pożar gwałtowny Występuje wtedy, gdy chmura mieszaniny palnego gazu i powietrza ulegnie zapłonowi. Kształt płomienia przybiera postać, jaką miała chmura mieszaniny przed zapłonem. Jego wielkość zależy również od miejsca zapłonu wewnątrz chmury. Prędkość spalania jest funkcją stężenia substancji palnej w chmurze i w mniejszym stopniu zależy również od prędkości wiatru. Jeżeli w chwili zapłonu chmura uwolnień obejmuje miejsce uwolnienia to, w zależności od sposobu uwolnienia, mogą zaistnieć warunki do powstania pożaru rozlewiska lub pożaru strumieniowego. Jest również możliwe, że front płomienia osiągnie wystarczająco dużą prędkość dla powstania wybuchu.

47
Pożar kulisty Kula ognista może powstać przy gwałtownych uwolnieniach, którym towarzyszy również gwałtowne mieszanie i zapłon. Początkowo (przed zapłonem) chmura przybiera postać półkuli. W wyniku termicznych sił wyporu, tuż po zapłonie, szybko zamienia się w kulę. Jeżeli wypływ substancji palnej jest skierowany bezpośrednio w górę, to kula ognista tworzy się natychmiastowo po zapłonie chmury. Ważnym źródłem kul ognistych jest zespół zjawisk zwany BLEVE (Boiling Liquid Expanding Vapour Explosion - eksplozja rozprężającej się pary wrzącej cieczy). Charakterystyczne są one dla palnych cieczy utrzymywanych pod ciśnieniem w temperaturze otoczenia. Zdarzenie zaczyna się od zewnętrznego pożaru, zasilanego ewentualnie przez substancję palną wypływającą ze zbiornika. Płomienie atakują powierzchnię zbiornika będącą w kontakcie z jego ciekłą zawartością. Wrzenie cieczy zwiększa ciśnienie pary, ale utrzymuje jednocześnie względnie chłodna zwilżaną część powierzchni zbiornika. Jednak w miejscach, gdzie ogień atakuje z zewnątrz, część powierzchni zbiornika kontaktuje się tylko z parą odbiór ciepła jest mały. W wyniku tego, w tych miejscach gwałtownie rośnie temperatura powierzchni metalu. Metalowa powłoka zbiornika słabnie i przy wzrastaniu ciśnienia w zbiorniku gwałtownie pęka. W wyniku uszkodzenia zbiornika, jego zawartość utrzymywana pod ciśnieniem zostaje gwałtownie wyrzucona na zewnątrz i rozprężając się tworzy chmurę pary z kropelkami cieczy. Płomienie na zewnątrz zbiornika powodują zapłon chmury i utworzenie się kuli ognistej olbrzymich rozmiarów ( nawet do 600 m średnicy ).
 chmura przybiera postać półkuli. W wyniku termicznych sił wyporu, tuż po zapłonie, szybko zamienia się w kulę. Jeżeli wypływ substancji palnej jest skierowany bezpośrednio w górę, to kula ognista tworzy się natychmiastowo po zapłonie chmury. Ważnym źródłem kul ognistych jest zespół zjawisk zwany BLEVE (Boiling Liquid Expanding Vapour Explosion - eksplozja rozprężającej się pary wrzącej cieczy). Charakterystyczne są one dla palnych cieczy utrzymywanych pod ciśnieniem w temperaturze otoczenia. Zdarzenie zaczyna się od zewnętrznego pożaru, zasilanego ewentualnie przez substancję palną wypływającą ze zbiornika. Płomienie atakują powierzchnię zbiornika będącą w kontakcie z jego ciekłą zawartością. Wrzenie cieczy zwiększa ciśnienie pary, ale utrzymuje jednocześnie względnie chłodna zwilżaną część powierzchni zbiornika. Jednak w miejscach, gdzie ogień atakuje z zewnątrz, część powierzchni zbiornika kontaktuje się tylko z parą odbiór ciepła jest mały. W wyniku tego, w tych miejscach gwałtownie rośnie temperatura powierzchni metalu. Metalowa powłoka zbiornika słabnie i przy wzrastaniu ciśnienia w zbiorniku gwałtownie pęka. W wyniku uszkodzenia zbiornika, jego zawartość utrzymywana pod ciśnieniem zostaje gwałtownie wyrzucona na zewnątrz i rozprężając się tworzy chmurę pary z kropelkami cieczy. Płomienie na zewnątrz zbiornika powodują zapłon chmury i utworzenie się kuli ognistej olbrzymich rozmiarów ( nawet do 600 m średnicy ).")
48
Przyczyny, czy też zdarzenia, które doprowadzają do wybuchu mogą być różne, najczęściej jest to:
samonagrzewanie się cieczy, ogrzanie cieczy w zbiorniku lub instalacji na skutek oddziaływania pożaru, gwałtowny wyciek podczas wypadków komunikacyjnych.

49
Powstawaniu Bleve towarzyszą zwykle efekty :
rozpryskiwanie spalające się cieczy, powstawanie fali uderzeniowej (wybuchowej ), powstawanie zjawiska o nazwie fireball ( płomień w kształcie kuli ), silne promieniowanie termiczne, powstawanie odłamków z części zbiorników, czy instalacji.
, powstawanie zjawiska o nazwie fireball ( płomień w kształcie kuli ), silne promieniowanie termiczne, powstawanie odłamków z części zbiorników, czy instalacji.")
50
Powstanie BLEVE w czasie katastrofy cysterny ze skroplonym propanem

51
BLEVE – tragedia w San Juanico (MEXICO CITY)
► ranek r. - zakład dystrybucji gazu LPG, ► gęsto zaludniony obszar w odległości 300m od zakładu, ► pękł rurociąg o średnicy 200 mm, ► operator nie mógł porozumieć się z terminalem, skąd można było odciąć wypływ gazu, ► po 10 min olbrzymia chmura par LPG zapaliła się, ► po następnych 5 min pożar spowodował wybuch BLEVE dwóch dużych zbiorników kulistych (każdy zawierał 1500 m3 skroplonego gazu), ► kule ogniste osiągały 600 m, ► fragmenty zbiorników i instalacji o wadze ton lądowały w odległości 100 do 890 m od miejsca wybuchu, ► 15 z 48 cylindrycznych zbiorników (20 ton każdy), zmieniło się w pociski wznoszące się na wysokość 100m, po czym lądowały w odległości m ► temperatura : w pobliżu zbiorników kulistych osiągnęła oC, w płomieniu – fireball 1200 oC, 200 m dalej – oC. ► na skutek pożaru i wybuchów spłonęło około m3 skroplonego gazu, ► zginęło 500 osób, 2000 odniosło poważne obrażenia (większość poparzenia), ► ludzi ewakuowano,
, ► kule ogniste osiągały 600 m, ► fragmenty zbiorników i instalacji o wadze ton lądowały w odległości 100 do 890 m od miejsca wybuchu, ► 15 z 48 cylindrycznych zbiorników (20 ton każdy), zmieniło się w pociski wznoszące się na wysokość 100m, po czym lądowały w odległości m. ► temperatura : w pobliżu zbiorników kulistych osiągnęła oC, w płomieniu – fireball 1200 oC, 200 m dalej – oC. ► na skutek pożaru i wybuchów spłonęło około m3 skroplonego gazu, ► zginęło 500 osób, 2000 odniosło poważne obrażenia (większość poparzenia), ► ludzi ewakuowano,")
52
WŁASNOŚCI WYBUCHOWE PALNYCH GAZÓW

53
Przebieg procesu palenia gazów i par
Przebieg spalania gazów zależy od tego, czy spala się „sam gaz” czy też jego mieszanina z powietrzem. Spalanie czystego gazu, np. u wylotu palnika , zależy od powierzchni otwory i od ciśnienia gazu u wylotu. Im ciśnienie będzie wyższe tym płomień będzie dłuższy i trudniejsza dyfuzja powietrza potrzebnego do spalania – spalanie jest niecałkowite , płomień świecący. Gdy z palnika wydobywa się gaz już zawierający pewną ilość powietrza, płomień jest krótszy i ma wyższą temperaturę -wykorzystywane jest to przy budowie palników np. kuchenek gazowych. Niecałkowite spalanie gazów jest bardzo niebezpieczne bowiem powstają znaczne ilości trującego tlenku węgla oraz istnieje niebezpieczeństwo tworzenia się mieszanin wybuchowych i ich wybuchów w miejscach odległych od płomienia, w dogodnych warunkach dyfuzji powietrza do spalin. Inny charakter ma spalanie mieszaniny gazu palnego z powietrzem, jeżeli oba czynniki są w odpowiednich proporcjach. Spalanie gazu przebiega wówczas z bardzo dużą szybkością, dochodzącą do kilkuset metrów na sekundę. Tak szybkie spalanie połączone jest z gwałtownym wzrostem ciśnienia. W mieszaninach gazowych reakcja palenia zachodzi w bardzo cienkiej warstwie palącej się substancji przylegającej bezpośrednio do frontu płomienia.

54
Frontem płomienia nazywa się granicę między płomieniem, czyli już spalonym paliwem a jeszcze nie spaloną substancją. W bardzo cienkiej warstwie pomiędzy spaloną i nie spaloną substancją następuje stopniowy wzrost temperatury od temperatury wyjściowej do temperatury płomienia i zmiana substancji w produkt spalania, czyli w gazy spalinowe. Tę przejściową warstwę ogrzewa ciepło płomienia. Ogrzanie przyspiesza reakcję chemiczną. Wydzielające się przy tym ciepło ogrzewa następną warstwę, która po ogrzaniu spala się itd. aż warstwa po warstwie spali się cała ilość substancji. Im szybsze jest wydzielanie się ciepła, tym szybszy jest przebieg reakcji spalania i tym szybciej rozprzestrzenia się płomień. Spalanie mieszanin gazowych przebiega z bardzo dużą szybkością w sposób wybuchowy. Mechanizm spalania gazów Chemiczne równania reakcji spalania gazów są to stechiometryczne równania spalania całkowitego gazów tzn. że w produktach spalania są dwutlenek węgla i woda. Okazuje się jednak, co potwierdzają wyniki badań, że reakcje spalania w płomieniu nie przebiegają jednoetapowo, lecz, że mechanizm spalania obejmuje serię następujących po sobie ,,kroków" w których biorą udział wysokoenergetyczne atomy i rodniki typu H, OH, HO2 i inne. Chociaż ich czas trwania w płomieniu jest bardzo krótki, są one odpowiedzialne za szybkie zużywanie się paliwa, a więc za szybkość spalania. Ich stężenie w płomieniu nie zmniejsza się w czasie, ponieważ atomy i rodniki ciągle regenerują się, w wyniku zachodzących w płomieniu reakcji łańcuchowych.

55
Reakcja łańcuchowa jest to więc typ reakcji wieloetapowej, przebiegającej w wyniku aktywizacji obojętnych cząsteczek przez reaktywne atomy. Najprostszym przykładem łańcuchowej reakcji spalania jest spalanie wodoru w tlenie, które przebiega m.in. wg następującego schematu: a) H2 + λ = 2H' b) H’ + O2 OH’ + O’ c) O’ + H2 OH’ + H’ d) OH’ + H2 H2O + H’ e) H’ + OH + M. = H2O (zapoczątkowanie reakcji a) pod wpływem światła; w wyniku powstają z obojętnej cząsteczki wodoru, dwa aktywne atomy H' (rodnik). b, c, d - reakcje rozwinięcia łańcucha; z jednego rodnika H' tworzą się dwa rodniki OH' + O. e - zakończenie reakcji w wyniku zetknięcia się rodnika z cząstką obojętną M
 H2 + λ = 2H b) H’ + O2 OH’ + O’ c) O’ + H2 OH’ + H’ d) OH’ + H2 H2O + H’ e) H’ + OH + M. = H2O. (zapoczątkowanie reakcji a) pod wpływem światła; w wyniku powstają z obojętnej cząsteczki wodoru, dwa aktywne atomy H (rodnik). b, c, d - reakcje rozwinięcia łańcucha; z jednego rodnika H tworzą się dwa rodniki OH + O. e - zakończenie reakcji w wyniku zetknięcia się rodnika z cząstką obojętną M.")
56
Przykłady reakcji łańcuchowej
Taki mechanizm spalania gazów ma bardzo istotne znaczenie przy przerywaniu tego procesu – likwidacja rodników przerywa proces palenia – inhibicyjne działanie proszku gaśniczego typu BC.

57
Palenie dyfuzyjne i kinetyczne
Reakcję spalania, której szybkość zależy od dyfuzji powietrza do paliwa nazywa się spalaniem dyfuzyjnym. Reakcję spalania mieszaniny palnej (substancja palna wstępnie zmieszana z utleniaczem) nazywa się spalaniem kinetycznym. Spalanie dyfuzyjne Palenie dyfuzyjne oparte na przykładzie gazu palnego – wodoru. Z chwilą zetknięcia się wodoru z powietrzem, a w następstwie różnicy ciśnień cząsteczkowych, zachodzi dyfuzja molekularna. W wyniku wzajemnego przenikania się cząsteczek wodoru, azotu i tlenu, tworzy się mieszanina. Stężenie wodoru w różnych punktach mieszaniny jest niejednakowe i zmniejsza się w miarę wzrostu odległości od miejsca dyfuzji wodoru, do powietrza. W zależności od stężenia wodoru całą objętość tworzącej się mieszaniny można podzielić na trzy części:
 nazywa się spalaniem kinetycznym. Spalanie dyfuzyjne. Palenie dyfuzyjne oparte na przykładzie gazu palnego – wodoru. Z chwilą zetknięcia się wodoru z powietrzem, a w następstwie różnicy ciśnień cząsteczkowych, zachodzi dyfuzja molekularna. W wyniku wzajemnego przenikania się cząsteczek wodoru, azotu i tlenu, tworzy się mieszanina. Stężenie wodoru w różnych punktach mieszaniny jest niejednakowe i zmniejsza się w miarę wzrostu odległości od miejsca dyfuzji wodoru, do powietrza. W zależności od stężenia wodoru całą objętość tworzącej się mieszaniny można podzielić na trzy części:")
58
Tworzenie się mieszaniny palnej w płomieniu w wyniku dyfuzji molekularnej.
1. obszar, w którym stężenie składnika palnego przewyższa górną granicę zapalności, 2. obszar stężeń w zakresie wybuchowym, 3. obszar, w którym stężenie składnika palnego jest niższe od dolnej granicy wybuchowości

59
W 1 obszarze znajduje się mieszanina zawierająca duży procent wodoru, a mało tlenu. Taka mieszanina nie jest zdolna do spalania się, ponieważ stężenie wodoru jest wyższe od stężenia odpowiadającego górnej granicy zapalności, czyli jest wyższe od maksymalnego stężenia, przy którym zachodzi spalanie. W obszarze 2- znajduje się mieszanina, w której stężenie wodoru leży w obszarze zapalności wodoru, a więc zdolna do zapalenia się. W 3- obszarze stężenie wodoru jest niższe od wartości dolnej granicy, tzn. jest niższe od minimalnego stężenia, przy którym już zachodzi spalanie, a więc mieszanina nie spala się. Z chwalą zbliżenia do powstałej mieszaniny źródła zapalenia, spalanie zachodzi tylko w obszarze 2. W czasie spalania się tej mieszaniny, tworzy się kosztem dyfuzji cząstek cienka warstewka mieszaniny, która warunkuje ciągłość spalania. Utrzymujący się w wyniku tej dyfuzji płomień, nazywa się jak wspomniano płomieniem dyfuzyjnym.

60
Podział i charakterystyka zjawisk towarzyszących spalaniu kinetycznemu
Spalenie kinetyczne Reakcję spalania mieszaniny palnej (substancja palna wstępnie zmieszana z utleniaczem) nazywa się spalaniem kinetycznym. W objętości wypełnionej mieszaniną palną płomień przemieszcza się z narastającą prędkością. Podział i charakterystyka zjawisk towarzyszących spalaniu kinetycznemu
 nazywa się spalaniem kinetycznym. W objętości wypełnionej mieszaniną palną płomień przemieszcza się z narastającą prędkością. Podział i charakterystyka zjawisk towarzyszących spalaniu kinetycznemu.")
61
Przemieszczanie się płomienia w mieszaninach gazowych może przebiegać na zasadzie deflagracji lub detonacji. Spalanie deflagracyjne związane jest z wymianą ciepła i masy w obszarze spalania. Znajdująca się przed frontem płomienia świeża mieszanina jest ogrzewana ciepłem wywiązującym się w strefie spalania. Wymiana ciepła następuje w wyniku przewodnictwa cieplnego, promieniowania i konwekcji. Jednocześnie do świeżej mieszaniny dyfundują wolne rodniki, a do strefy spalania cząsteczki świeżej mieszaniny palnej. Przy spalaniu deflagracyjnym płomień może mieć charakter laminarny lub turbulentny. Spalanie deflagracyjne przebiega z prędkościami mieszczącymi się w zakresie od kilku cm/s do prędkości dźwięku. Spalanie detonacyjne przenosi się ze stałą prędkością naddźwiękową mieszczącą się w granicach 1400 = 3000 m/s. Detonacja w mieszaninach węglowodorów z powietrzem przemieszcza się z prędkością 1600 m/s. Wybuchy heterogeniczne charakteryzują się rozdzielaniem strefy reakcji od strefy nie reagującej poprzez front reakcji (płomień), który przemieszcza się przez mieszaninę palną z określoną prędkością. Jeśli prędkość ta jest mniejsza niż prędkość dźwięku, to wybuch ten jest nazywany deflagracją, a mechanizm propagacji oparty jest o wymianę ciepła
, który przemieszcza się przez mieszaninę palną z określoną prędkością. Jeśli prędkość ta jest mniejsza niż prędkość dźwięku, to wybuch ten jest nazywany deflagracją, a mechanizm propagacji oparty jest o wymianę ciepła.")
62
Budowę płomienia dyfuzyjnego przedstawia rysunek poniżej.
W strefie gazów następuje przygotowanie materiału palnego do spalania tzn. ogrzewanie, dysocjacja termiczna i częściowe utlenianie powstałych produktów rozkładu, wskutek dyfuzji tlenu ze strefy spalania. Strefa spalania jest to bardzo cienka warstwa płomienia, w której powstaje i spala się mieszanina substancji palnej z powietrzem. Strefa mieszania się produktów spalania z powietrzem Strefa spalania Strefa palnych gazów Budowa płomienia dyfuzyjnego

63
Typowe prędkości przemieszczania się płomienia zmieniają się w szerokich granicach od 10 do 100 m/s i proces jest stosunkowo wolny. Ponieważ mechanizm propagacji płomienia jest identyczny do tego jaki występuje w pożarze, stąd deflagracja zwana jest również "wybuchowym spalaniem". Przemieszczający się front reakcji i wydzielające się ciepło powodują powstawanie przed frontem fali ciśnienia, która przemieszcza się z prędkością poddźwiękową. Wzrost ciśnienia jest stosunkowo niewielki i może dochodzić do 7 = 10 barów (dla zamkniętych przestrzeni) natomiast temperatura płomienia dochodzi do 2000, a nawet 3000 K (spalanie metali). Fale ciśnienia wykorzystywane są jako sygnały aktywujące urządzenia przeciwwybuchowe do dławienia inicjacji wybuchu. Szybkość propagacji płomienia przez gazową mieszaninę palną jest uwarunkowana głównie jej reaktywnością i warunkami początkowymi (ciśnienia i temperatury). Wpływ tych czynników jest opisany przy omawianiu granic wybuchowości. Jeśli w obszarze zagrożonym znajduje się zwiększona ilość tlenu (np. nieszczelności butli tlenowych w szpitalach, przy procesach technologicznych, itp.) należy liczyć się ze wzrostem zagrożenia, ze względu na wzrost szybkości propagacji spalania. Innym, istotnym czynnikiem dla szybkości propagacji spalania w mieszaninach gazowych jest turbulencja, której obecność w gazach nie spalonych powoduje wzrost szybkości propagacji płomienia. Ze wzrostem powierzchni pożaru pojawia się turbulencja wywołana własnym, nie wymuszonym transportem masy (średnica pożaru > 0.3 m). Turbulencja wzmaga gwałtowność pożaru i wybuchu i odgrywa znaczącą rolę w szybkości jego rozwoju.
. Wpływ tych czynników jest opisany przy omawianiu granic wybuchowości. Jeśli w obszarze zagrożonym znajduje się zwiększona ilość tlenu (np. nieszczelności butli tlenowych w szpitalach, przy procesach technologicznych, itp.) należy liczyć się ze wzrostem zagrożenia, ze względu na wzrost szybkości propagacji spalania. Innym, istotnym czynnikiem dla szybkości propagacji spalania w mieszaninach gazowych jest turbulencja, której obecność w gazach nie spalonych powoduje wzrost szybkości propagacji płomienia. Ze wzrostem powierzchni pożaru pojawia się turbulencja wywołana własnym, nie wymuszonym transportem masy (średnica pożaru > 0.3 m). Turbulencja wzmaga gwałtowność pożaru i wybuchu i odgrywa znaczącą rolę w szybkości jego rozwoju.")
64
Geometria układu może być czynnikiem znacznie zwiększającym szybkość propagacji spalania. W przestrzeniach zamkniętych lub ograniczonych z dużą ilością przegród i otworów (np. elementów urządzeń i instalacji, drzwi itp.) propagujący proces spalania będzie wywoływał własną turbulencję przyśpieszającą spalanie. Gwałtowny wzrost ciśnienia przy zapłonie mieszaniny palnej od strony zamkniętego końca rurociągu lub tunelu może w pewnych warunkach spowodować gwałtowny wzrost szybkości propagacji płomienia i wywołać detonację, tzn. proces, w którym fala uderzeniowa i płomień poruszają się z tą samą szybkością, większą od szybkości dźwięku. Wzrost długości tunelu (w praktyce istotny jest stosunek długości do średnicy) oraz istnienie przegród i przewężeń sprzyjają przejściu ze spalania deflagracyjnego do detonacji. Detonacja Detonacja może powstać w mieszaninach palnych gazów i par z powietrzem lub z tlenem o stężeniu mieszczącym się w przedziałach węższych niż granice wybuchowości. W czasie wybuchu detonacyjnego, strefa reakcji przemieszcza się z prędkością od m/s, a dla cieczy, czy ciał stałych nawet może osiągnąć 8000 m/s. Mechanizm polega na gwałtownym adiabatycznym sprężaniu występującym w fali uderzeniowej, co powoduje ogrzanie reagentów powyżej ich temperatury zapłonu powodując dalsze spalanie. Uwolnione ciepło służy do dalszej propagacji procesu wybuchu (sprężanie uderzeniowe). W detonacji zachodzącej w bardzo krótkim czasie nie występuje zjawisko wyprzedzania strefy reakcji przez falę ciśnienia, stąd nie ma możliwości użycia tej fali jako sygnału ostrzegającego o nadchodzącym zagrożeniu.
. W detonacji zachodzącej w bardzo krótkim czasie nie występuje zjawisko wyprzedzania strefy reakcji przez falę ciśnienia, stąd nie ma możliwości użycia tej fali jako sygnału ostrzegającego o nadchodzącym zagrożeniu.")
65
Przed nadejściem frontu fali uderzeniowej ciśnienie jest na poziomie ciśnienia atmosferycznego. Z chwilą nadejścia frontu ciśnienie gwałtownie rośnie, aż do wartości maksymalnej, zwanej szczytowym dodatnim nadciśnieniem. Następnie ciśnienie opada do wartości ciśnienia atmosferycznego w okresie czasu zwanym okresem fazy dodatniej. Okres dalszego spadku ciśnienia i ewentualnego jego powrotu do ciśnienia atmosferycznego nazywamy okresem fazy ujemnej. Ważnymi parametrami całego procesu są: wartość maksymalna nadciśnienia i pole pod funkcją opisującą zależności ciśnienia od czasu w okresie fazy dodatniej (rysunek poniżej). O charakterze i mechanizmie wybuchu decydują liczne parametry, które można zestawić w cztery zasadnicze grupy: 1. właściwości materiału (fizyczne, chemiczne, stabilność, ciepło spalania etc.), 2. charakterystyka przestrzeni, w której zachodzi spalanie (rozmiar, przeszkody ograniczone czy otwarte, etc.), 3. właściwości mieszaniny wybuchowej (stan skupienia, stężenie, turbulencja, ciśnienie i temperatura, obecność gazu inertnego, mieszanina hybrydowa), 4. czynniki zapłonu (energia, temperatura).
, 2. charakterystyka przestrzeni, w której zachodzi spalanie (rozmiar, przeszkody ograniczone czy otwarte, etc.), 3. właściwości mieszaniny wybuchowej (stan skupienia, stężenie, turbulencja, ciśnienie i temperatura, obecność gazu inertnego, mieszanina hybrydowa), 4. czynniki zapłonu (energia, temperatura).")
66
Wyidealizowana struktura fali uderzeniowej

67
Zjawiska zwane powszechnie wybuchami można podzielić – w oparciu o ich przyczyny – na pięć grup:
wybuchy powstałe w wyniku uwalniania energii, czego przyczyną jest szybko przebiegająca reakcja utleniania (spalanie mieszaniny utworzonej z powietrza i gazów albo par węglowodorów), wybuchy wywołane szybkim rozkładem niektórych ciał (rozkład saletry amonowej albo acetylenu), wybuchy powstałe w następstwie nadmiernego ciśnienia wewnętrznego (wybuchy kotłów lub aparatów ze sprężonym gazem) albo osłabienia ścianek naczyń zawierających gazy pod ciśnieniem (działanie płomienia na część „nie zwilżoną” butli gazowej), 4. wybuchy wywołane przez gwałtowne uwolnienie energii w przebiegu syntezy jądrowej (fuzji) lub reakcji rozszczepienia jądrowego – bomba A (atomowa) albo H (wodorowa), wybuchy spowodowane przez niekontrolowane reakcje polimeryzacji, którym towarzyszy szybkie uwalnianie energii (samorzutna polimeryzacja nadtlenków organicznych).
, wybuchy wywołane szybkim rozkładem niektórych ciał (rozkład saletry amonowej albo acetylenu), 3. wybuchy powstałe w następstwie nadmiernego ciśnienia wewnętrznego (wybuchy kotłów lub aparatów ze sprężonym gazem) albo osłabienia ścianek naczyń zawierających gazy pod ciśnieniem (działanie płomienia na część „nie zwilżoną butli gazowej), 4. wybuchy wywołane przez gwałtowne uwolnienie energii w przebiegu syntezy jądrowej (fuzji) lub reakcji rozszczepienia jądrowego – bomba A (atomowa) albo H (wodorowa), 5. wybuchy spowodowane przez niekontrolowane reakcje polimeryzacji, którym towarzyszy szybkie uwalnianie energii (samorzutna polimeryzacja nadtlenków organicznych).")
68
Analiza powyższych definicji prowadzi do stwierdzenia, że zjawiska, które określa się wybuchem można podzielić ze względu na przyczynę ich powstania na wybuch fizyczny, chemiczny i jądrowy. Do wybuchów fizycznych zaliczamy: gwałtowne rozerwanie kotła parowego, rozsadzenie zbiornika przez sprężony gaz, wyładowania atmosferyczne, erupcje wulkaniczne, wyrzuty cieczy palnych w czasie pożarów zbiorników, przekraczanie bariery dźwięku przez samoloty naddźwiękowe itp. zjawiska. Wybuchy fizyczne nie są powszechnie wykorzystywane w praktyce pożarniczej. Mają one raczej charakter działań szkodliwych –destrukcyjnych, dlatego są przedmiotem specjalnych badań naukowych. Warto jednak wspomnieć o pozytywnym wykorzystaniu wybuchów fizycznych w rurach uderzeniowych, których zastosowanie utorowało, między innymi, drogę badaniom zjawisk towarzyszących procesowi wejścia pojazdu kosmicznego w atmosferę. Do wybuchów chemicznych zalicza się te wszystkie wybuchy, których przyczyną są reakcje chemiczne. Analiza podanych powyżej przykładów wskazuje na różnorodność przyczyn chemicznych prowadzących do wybuchu: utlenianie, rozkład termiczny, polimeryzacja. Wybuchem chemicznym nazwiemy szybkie, egzotermiczne procesy chemiczne zachodzące w wybuchowych mieszaninach gazowych i pyłowo-powietrznych oraz w stałych i ciekłych materiałach wybuchowych. W naukowym słownictwie francuskim słowo wybuch (explosion) oznacza zarówno zjawisko przebiegające powoli, znane pod nazwą deflagracji, jak również zjawisko przebiegające szybko, któremu towarzyszy gwałtowny huk i które określa się nazwą detonacji; w pierwszym przypadku szybkość rozprzestrzeniania się płomienia nigdy nie przekracza kilku metrów na sekundę, natomiast w drugim przypadku szybkość ta może osiągnąć kilka kilometrów na sekundę.
 oznacza zarówno zjawisko przebiegające powoli, znane pod nazwą deflagracji, jak również zjawisko przebiegające szybko, któremu towarzyszy gwałtowny huk i które określa się nazwą detonacji; w pierwszym przypadku szybkość rozprzestrzeniania się płomienia nigdy nie przekracza kilku metrów na sekundę, natomiast w drugim przypadku szybkość ta może osiągnąć kilka kilometrów na sekundę.")
69
Klasyfikacja wybuchów według sposobu wywiązywanej energii
Istnieje wiele procesów, które mogą prowadzić do wybuchów. Przedstawiona poniżej tabela - podaje jedną z prób ich klasyfikacji według rodzaju wywiązanej energii. Zawiera ona przykłady wybuchów naturalnych, wybuchów zamierzonych oraz wybuchów przypadkowych. Klasyfikacja wybuchów według sposobu wywiązywanej energii

70
Klasyfikacja wybuchów

71
Czynniki wpływające na wybuch

72
Granice wybuchowości Spalanie mieszanin zachodzi w ściśle określonych granicach składu i związane jest z bilansem energetycznym w obszarze reakcji. Istotnym czynnikiem warunkującym istnienie granic wybuchowości jest stosunek szybkości wyzwalania ciepła w wyniku reakcji spalania do szybkości jego odprowadzania do otoczenia. Zapłon i dalsze samoczynne przemieszczanie się płomienia w mieszaninach gazowych jest możliwe w pewnym zakresie stężeń obu składników reakcji spalania. Zakres ten jest określony dolną i górną granicą wybuchowości (DGW i GGW). Dolna granica wybuchowości (DGW) – jest to najniższe stężenie paliwa w mieszaninie palnej, poniżej którego nie jest możliwy jej zapłon pod wpływem czynnika inicjującego ( bodźca energetycznego), w określonych warunkach badania. Górna granica wybuchowości (GGW) – jest to najwyższe stężenie paliwa w mieszaninie palnej, powyżej którego nie jest możliwy jej zapłon pod wpływem czynnika inicjującego (bodźca energetycznego), w określonych warunkach badania.
. Dolna granica wybuchowości (DGW) – jest to najniższe stężenie paliwa w mieszaninie palnej, poniżej którego nie jest możliwy jej zapłon pod wpływem czynnika inicjującego ( bodźca energetycznego), w określonych warunkach badania. Górna granica wybuchowości (GGW) – jest to najwyższe stężenie paliwa w mieszaninie palnej, powyżej którego nie jest możliwy jej zapłon pod wpływem czynnika inicjującego (bodźca energetycznego), w określonych warunkach badania.")
73
Mieszaniny gazów palnych z powietrzem
Jeżeli na odcinku AB (rysunek poniżej) wyobrazimy sobie wszystkie możliwe składy mieszaniny gazu palnego z powietrzem, wówczas otrzymamy trzy charakterystyczne przedziały stężeń (1, 2 i 3). 1 Mieszaniny ubogie 3 Mieszaniny bogate 2 Mieszaniny wybuchowe i zapalne Mieszaniny gazów palnych z powietrzem
 wyobrazimy sobie wszystkie możliwe składy mieszaniny gazu palnego z powietrzem, wówczas otrzymamy trzy charakterystyczne przedziały stężeń (1, 2 i 3). 1 Mieszaniny ubogie. 3 Mieszaniny bogate. 2 Mieszaniny wybuchowe i zapalne. Mieszaniny gazów palnych z powietrzem.")
74
obszar A - DGW: przedstawiający mieszaniny ubogie w składnik palny, zawierające znaczny nadmiar tlenu, niepalne, obszar DGW - GGW: obejmujący mieszaniny palne i wybuchowe, w których płomień przemieszcza się z różnymi prędkościami, obszar GGW - B: zawierający mieszaniny bogate w składnik palny, mające znaczny niedomiar tlenu, niepalne, nie wybuchające. W przedziale mieszanin wybuchowych znajduje się punkt odpowiadający stężeniu stechiometrycznemu mieszaniny, w której stosunek obu substratów odpowiada teoretycznemu równaniu całkowitego spalania. Stężenie stechiometryczne związku organicznego, zawierającego x atomów węgla, y atomów wodoru i z atomów tlenu, w mieszaninie z tlenem t (% objętości) oblicza się ze wzoru: Ten sam wzór dla mieszanin z powietrzem
 oblicza się ze wzoru: Ten sam wzór dla mieszanin z powietrzem.")
75
Skład chemiczny paliwa ma istotny wpływ na granice wybuchowości, np
Skład chemiczny paliwa ma istotny wpływ na granice wybuchowości, np. w mieszaninach z powietrzem acetylen wybucha w szerokich granicach składu: od 2,3% obj. (DGW) do 82% obj. (GGW). Metan z powietrzem tworzy mieszaniny wybuchowe w znacznie węższych granicach składu: od 4,9% obj. (DGW) do 15,4% obj. (GGW). Zmiana utleniacza również powoduje istotne zmiany w granicach wybuchowości. Zmiany stężenia tlenu w mieszaninie silnie zmieniają zakres wybuchowości. Zależność tę przedstawia na przykładzie metanu rysunek poniżej. Podczas wzbogacania mieszaniny trójskładnikowej CH4/N2/02 w tlen, dolna granica wybuchowości praktycznie nie zmienia się do punktu A, ponieważ i tak tlen znajduje się w nadmiarze. W mieszaninach, w pobliżu górnej granicy wybuchowości mamy niedobór tlenu i każda zmiana jego stężenia silnie wpływa na przemieszczenie się granicy: wzrost stężenia tlenu powoduje podwyższenie górnej granicy wybuchowości, a spadek - jej obniżenie, aż do punktu A, gdzie obie granice schodzą się.
 do 82% obj. (GGW). Metan z powietrzem tworzy mieszaniny wybuchowe w znacznie węższych granicach składu: od 4,9% obj. (DGW) do 15,4% obj. (GGW). Zmiana utleniacza również powoduje istotne zmiany w granicach wybuchowości. Zmiany stężenia tlenu w mieszaninie silnie zmieniają zakres wybuchowości. Zależność tę przedstawia na przykładzie metanu rysunek poniżej. Podczas wzbogacania mieszaniny trójskładnikowej CH4/N2/02 w tlen, dolna granica wybuchowości praktycznie nie zmienia się do punktu A, ponieważ i tak tlen znajduje się w nadmiarze. W mieszaninach, w pobliżu górnej granicy wybuchowości mamy niedobór tlenu i każda zmiana jego stężenia silnie wpływa na przemieszczenie się granicy: wzrost stężenia tlenu powoduje podwyższenie górnej granicy wybuchowości, a spadek - jej obniżenie, aż do punktu A, gdzie obie granice schodzą się.")
76
Zależność granic wybuchowości metanu od stężenia tlenu

77
Granice zapalności mieszanin palnych par i gazów z powietrzem, oznacza się w % objętościowych, rzadziej w g/m3. Granice wybuchowości są zmienne i zależne od: ciśnienia - w miarę obniżania się ciśnienia, zakres granic zapalności mieszanin zwęża się aż do zrównania, dolnej i górnej granicy zapalności, czyli dla każdej mieszaniny gazów istnieje pewne krytyczne ciśnienie, poniżej którego własności wybuchowe i zdolność do zapalenia się przestają istnieć. Natomiast, gdy ciśnienie rośnie, granice zapalności zdążają asymptotycznie do określonych wartości lub w wielu przypadkach nie zmieniają się. Zmianę granic zapalności w zależności od zmiennego ciśnienia, wytłumaczyć można zmianą szybkości rozprzestrzeniania się płomienia. Rozprzestrzenianie się płomienia na całą objętość mieszaniny może zaistnieć tylko w tym przypadku, kiedy od zapalanej warstwy gazu do jeszcze nie zapalonej warstwy gazu przekazana będzie odpowiednia ilość ciepła. Rozprzestrzenianie się płomienia w mieszaninie palnej zachodzi z określą szybkością w stronę nieopalonej warstwy gazu, stykającej się bezpośrednio z czołem płomienia. Szybkość rozprzestrzeniania się płomienia w mieszaninie gazowej, w której stężenie składnika palnego odpowiada stężeniu dolnej lub górnej granicy zapalności, ma minimalną wielkość. Przy zawartości składnika palnego w mieszaninie poza obszarem zapalności, rozprzestrzenianie płomienia na całą objętość mieszaniny staje się niemożliwa;

78
Zmiana , pod wpływem ciśnienia, granic wybuchowości mieszaniny metanu z powietrzem
Granice wybuchowości tlenku węgla w mieszaninie z powietrzem podczas zmniejszania ciśnienia

79
b) temperatury - w miarę wzrostu temperatury mieszaniny palnej gazów, granice zapalności rozszerzają się, czyli mieszanina staje się bardziej zapalna; Wpływ temperatury na granice wybuchowości mieszanin gazowych pod stałym ciśnieniem

80
c) ilości gazu obojętnego w mieszaninie np
c) ilości gazu obojętnego w mieszaninie np. dodanie dwutlenku węgla lub azotu do palnej mieszaniny, znacznie zmniejsza zakres granic zapalności; Zależność granic wybuchowości metanu od stężenia tlenu
 ilości gazu obojętnego w mieszaninie np. dodanie dwutlenku węgla lub azotu do palnej mieszaniny, znacznie zmniejsza zakres granic zapalności; Zależność granic wybuchowości metanu od stężenia tlenu.")
81
d) składu - najbardziej niebezpieczną i wybuchową nie jest ta mieszanina, której skład chemiczny pozwala na całkowite spalenie (skład stechiometryczny), lecz taka, która ma pewną nadwyżkę łatwozapalnej pary lub gazu, w stosunku do ilości tlenu zawartego w powietrzu;
 składu - najbardziej niebezpieczną i wybuchową nie jest ta mieszanina, której skład chemiczny pozwala na całkowite spalenie (skład stechiometryczny), lecz taka, która ma pewną nadwyżkę łatwozapalnej pary lub gazu, w stosunku do ilości tlenu zawartego w powietrzu;")
82
e) miejsca zainicjowania zapłonu i kierunku dalszego rozprzestrzeniania się płomienia. Przy zapłonie górnym w rurze pionowej, istnieją dla mieszaniny warunki najbardziej niesprzyjające w przekazywaniu ciepła, ponieważ ciepło nie jest przenoszone na drodze konwekcji; Zmiany granic wybuchowości par i gazów w mieszaninie z powietrzem przy rozprzestrzenianiu się płomienia z dołu do góry (↑) z góry do dołu (↓) , prostopadle (→)
 z góry do dołu (↓) , prostopadle (→)")
83
f) stężenia tlenu w mieszaninie (bardzo znaczny wpływ na wartość górnej granicy zapalności). Przy górnej granicy zapalności tlen znajduje się w niedomiarze. O ile w takiej mieszaninie w miejsce azotu wprowadzi się tlen, wówczas większa część składnika palnego będzie mogła zareagować i stężenie składnika palnego odpowiadające górnej granicy zapalności ulegnie podniesieniu. W mieszaninach odpowiadających dolnej granicy zapalności, tlen znajduje się w nadmiarze, czyli zwiększenie stężenia tlenu wywiera minimalny wpływ na obniżenie wartości liczbowej dolnej granicy zapalności. g) bodźca termicznego - w miarę zwiększania się mocy początkowego impulsu, granice zapalności również rozszerzają się
 bodźca termicznego - w miarę zwiększania się mocy początkowego impulsu, granice zapalności również rozszerzają się.")
84
Wykorzystanie granic wybuchowości w praktyce
Oznaczanie granic wybuchowości gazów i par cieczy w praktyce pozwala na ocenę zagrożenia wybuchem pomieszczeń oraz przestrzeni zewnętrznych i w konsekwencji wyznaczenie w pomieszczeniach i przestrzeniach zewnętrznych odpowiednich stref zagrożenia wybuchem. Na podstawie § Rozporządzenia Ministra Spraw Wewnętrznych i Administracji z dnia 16 czerwca 2003 roku - w sprawie ochrony przeciwpożarowej budynków, innych obiektów budowlanych i terenów Dziennik Ustaw Nr 121 pozycja 1138 – w obiektach i na terenach przyległych, gdzie prowadzone są procesy technologiczne z użyciem materiałów mogących wytworzyć mieszaniny wybuchowe lub w których materiały takie są magazynowane, powinna być dokonana ocena zagrożenia wybuchem. § Ocena , o której mowa w ust. 1, obejmuje wskazanie pomieszczeń zagrożonych wybuchem, wyznaczenie w pomieszczeniach i przestrzeniach zewnętrznych odpowiednich stref zagrożenia wybuchem oraz wskazanie czynników mogących zainicjować zapłon. § Klasyfikację stref zagrożenia wybuchem określa Polska Norma dotycząca zapobiegania wybuchowi i ochronie przed wybuchem.

85
§ Pomieszczenie , w którym może wytworzyć się mieszanina wybuchowa, powstała z wydzielającej się takiej ilości palnych gazów, par, mgieł lub pyłów, której wybuch mógłby spowodować przyrost ciśnienia w tym pomieszczeniu przekraczający 5 kPa, określa się jako pomieszczenie zagrożone wybuchem. § W pomieszczeniu należy wyznaczyć strefę zagrożenia wybuchem, jeżeli może w nim występować mieszanina wybuchowa o objętości co najmniej 0,01 m3. Na podstawie § 41. tego rozporządzenia – strefy zagrożenia wybuchem wyznaczone w pomieszczeniach i przestrzeniach zewnętrznych , zakwalifikowane przed wejściem w życie rozporządzenia, klasyfikuje się odpowiednio, z uwzględnieniem wymagań określonych w § 33 ust.4, jako: 1) strefę Z strefa 0; 2) strefę Z strefa 1; 3) strefę Z strefa 2; 4) strefa Z strefa 20; 5) strefa Z strefa 21 lub strefa 22 ( w zależności od prawdopodobieństwa wystąpienia atmosfery wybuchowej).
 strefę Z 0 - strefa 0; 2) strefę Z 1 - strefa 1; 3) strefę Z 2 - strefa 2; 4) strefa Z 10 - strefa 20; 5) strefa Z 11 - strefa 21 lub strefa 22 ( w zależności od prawdopodobieństwa wystąpienia atmosfery wybuchowej).")
86
Z 0 – strefa , w której mieszania wybuchowa gazów, par lub mgieł występuje stale lub długotrwale w normalnych warunkach pracy. Z 1 – strefa, w której mieszanina wybuchowa gazów, par lub mgieł może występować w normalnych warunkach pracy. Z 2 – strefa, w której istnieje niewielkie prawdopodobieństwo wystąpienia mieszaniny wybuchowej gazów, par lub mgieł przy czym mieszanina wybuchowa może występować jedynie krótkotrwale.

87
Bodziec termiczny Funkcję źródła zapłonu mogą spełniać różne postacie energii cieplnej i różne przemiany energetyczne, których wspólną cechą jest wytworzenie odpowiednio wysokiej temperatury. Temperatury zapalenia gazów mieszczą się na ogół w zakresie C. Wyjątki stanowią: m.in. acetylen (czysty, bez domieszek) C, skroplony propan-butan 3200C. Ponieważ zapalenie jako proces jest określony bilansem ciepła tzn. ilością ciepła, które gromadzi się w układzie palnym i ilością ciepła, które jest tracone z układu, temperatury zapalenia gazów podane w tablicach dotyczą równoważnikowych stężeń gazów tzn. takich mieszanin, gdzie nie ma nadmiaru ani gazu ani powietrza. Po prostu wszystko się spala, są minimalne straty ciepła, gaz zapala się w najniższych temperaturach. Temperatury zapalenia nie są wielkościami stałymi. Zalezą one od m.in. składu mieszaniny, stężenia tlenu w cząsteczce palnej gazu i w otoczeniu, ciśnienia, katalizatorów itp. Przykładowe temperatury zapalenia gazów
 3050C, skroplony propan-butan 3200C. Ponieważ zapalenie jako proces jest określony bilansem ciepła tzn. ilością ciepła, które gromadzi się w układzie palnym i ilością ciepła, które jest tracone z układu, temperatury zapalenia gazów podane w tablicach dotyczą równoważnikowych stężeń gazów tzn. takich mieszanin, gdzie nie ma nadmiaru ani gazu ani powietrza. Po prostu wszystko się spala, są minimalne straty ciepła, gaz zapala się w najniższych temperaturach. Temperatury zapalenia nie są wielkościami stałymi. Zalezą one od m.in. składu mieszaniny, stężenia tlenu w cząsteczce palnej gazu i w otoczeniu, ciśnienia, katalizatorów itp. Przykładowe temperatury zapalenia gazów.")
89
Domieszki obojętnych gazów i par
Jeżeli do mieszaniny składnika palnego i utleniacza dodać gazu obojętnego wówczas następuje podwyższenie dolnej granicy wybuchowości i znaczne obniżenie górnej granicy wybuchowości. Obszar wybuchowości ulega zawężeniu i powyżej pewnej ilości danego składnika palnego mieszanina przestaje być palna. Dodatek gazu obojętnego (N2, CO2, H2O) z jednej strony zmniejsza ogólne stężenie tlenu w mieszaninie, zaś z drugiej strony zwiększa ilość bezproduktywnie ogrzewanego ciepłem reakcji gazu obojętnego. Większa ilość ciepła zostaje przekazana do gazu obojętnego, maleje temperatura spalania i rozprzestrzenianie się płomienia możliwe jest w zawężonym obszarze stężeń. Minimalne stężenie tlenu, przy którym możliwe jest jeszcze rozprzestrzenianie się płomienia przy rozcieńczeniu gazami obojętnymi dla większości substancji palnych ( z wyjątkiem H2, C2H2, CS2, C2H4, N2H4 ) wynosi zwykłe 10 – 16 % np. dla N2 około 11 % dla CO2 około 13 – 14 %, dla H2O około 16%. Obszar wybuchowości zanika , jeżeli ilość gazu obojętnego dodanego do mieszaniny większości substancji palnych w powietrzu wynosi od 20 – 40% np. dla N2 około 40%, dla CO2 około 28 – 30%, dla H2O około 20 – 23%. Rozcieńczenie mieszanin gazów i par palnych gazami obojętnymi ma duże znaczenie praktyczne w technologii i w prewencji pożarowej, umożliwia bowiem operowanie nimi w obecności źródeł zapłonu. Rozwiązania stosowane w praktyce polegają na wytwarzaniu atmosfery ochronnej.
 z jednej strony zmniejsza ogólne stężenie tlenu w mieszaninie, zaś z drugiej strony zwiększa ilość bezproduktywnie ogrzewanego ciepłem reakcji gazu obojętnego. Większa ilość ciepła zostaje przekazana do gazu obojętnego, maleje temperatura spalania i rozprzestrzenianie się płomienia możliwe jest w zawężonym obszarze stężeń. Minimalne stężenie tlenu, przy którym możliwe jest jeszcze rozprzestrzenianie się płomienia przy rozcieńczeniu gazami obojętnymi dla większości substancji palnych ( z wyjątkiem H2, C2H2, CS2, C2H4, N2H4 ) wynosi zwykłe 10 – 16 % np. dla N2 około 11 % dla CO2 około 13 – 14 %, dla H2O około 16%. Obszar wybuchowości zanika , jeżeli ilość gazu obojętnego dodanego do mieszaniny większości substancji palnych w powietrzu wynosi od 20 – 40% np. dla N2 około 40%, dla CO2 około 28 – 30%, dla H2O około 20 – 23%. Rozcieńczenie mieszanin gazów i par palnych gazami obojętnymi ma duże znaczenie praktyczne w technologii i w prewencji pożarowej, umożliwia bowiem operowanie nimi w obecności źródeł zapłonu. Rozwiązania stosowane w praktyce polegają na wytwarzaniu atmosfery ochronnej.")
90
Temperatura mieszaniny wybuchowej
Ze wzrostem temperatury granice wybuchowości "rozszerzają się". Jeśli mieszanina palna zostanie podgrzana, to zapłon może nastąpić przy stężeniu niższym niż DGW (oznaczonym dla mieszaniny o temperaturze pokojowej). Jest to bardzo ważna informacja praktyczna. Wydzielanie palnych par i gazów może nastąpić w sąsiedztwie rozgrzanych powierzchni. Wszędzie tam, gdzie temperatura mieszaniny wzrośnie, wybuch może nastąpić przy stężeniach niższych od wartości DGW, oznaczonych dla temperatury pokojowej. Wpływ temperatury na granice wybuchowości mieszanin gazowych pod stałym ciśnieniem
. Jest to bardzo ważna informacja praktyczna. Wydzielanie palnych par i gazów może nastąpić w sąsiedztwie rozgrzanych powierzchni. Wszędzie tam, gdzie temperatura mieszaniny wzrośnie, wybuch może nastąpić przy stężeniach niższych od wartości DGW, oznaczonych dla temperatury pokojowej. Wpływ temperatury na granice wybuchowości mieszanin gazowych pod stałym ciśnieniem.")
91
Kierunek rozprzestrzenianie się płomienia w mieszaninach gazowych
Położenie punktu powodującego zapalenie mieszaniny ma wpływ na wielkości granic wybuchowości. Oznacza to, że granice wybuchowości zmieniają się w zależności od tego, jak następuje rozprzestrzenianie się płomienia: z góry w dół z dołu do góry, czy też prostopadle. Zmiany granic wybuchowości par i gazów w mieszaninie z powietrzem przy rozprzestrzenianiu się płomienia z dołu do góry (↑) z góry do dołu (↓) , prostopadle (→)
 z góry do dołu (↓) , prostopadle (→)")
92
Jak z danych wynika , dolna granica wybuchowości jest najniższa przy rozprzestrzenianiu się płomienia z dołu do góry. Przy zapłonie dolnym w rurze pionowej, istnieją dla mieszaniny warunki najbardziej sprzyjające w przekazywaniu ciepła, ponieważ ciepło jest przenoszone na drodze konwekcji. Przy prostopadłym rozprzestrzenianiu się płomienia dolna granica wybuchowości dla wszystkich mieszanin ma wartość pośrednią pomiędzy obydwoma pierwszymi wielkościami. Górna granica wybuchowości dla wszystkich mieszanin jest największa przy rozprzestrzenianiu się płomienia z dołu do góry, najmniejsza przy rozprzestrzenianiu się płomienia z góry w dół. Przy prostopadłym rozprzestrzenianiu się płomienia górna granica wybuchowości ma wartość średnią dwóch pierwszych wielkości. Maksymalną szybkość rozprzestrzeniania się płomienia podczas spalania mieszanin uzyskuje się nie przy mieszaninach stechiometrycznych, lecz przy mieszaninach z nadmiarem palnego gazu.

93
Klasyfikacja pożarowo - wybuchowa gazów i par cieczy palnych
Kryterium podziału gazów na poszczególne grupy, jest stopień niebezpieczeństwa pożarowego, wynikający ze stopnia ich zdolności do wybuchu i stopnia palności. Gazy kwalifikuje się w cztery grupy: I grupa - gazy , tworzące mieszaniny wybuchowe z powietrzem przy stężeniu do % objętości, do tej grupy zalicza się między innymi: acetylen techniczny rozpuszczony, chlorek etylu, chlorek metylu, wodór , itp. II grupa – gazy, tworzące mieszaniny wybuchowe z powietrzem przy stężeniu powyżej 10 % objętości do których można zaliczyć między innymi amoniak, bromek metylu , itp. III grupa – gazy niepalne, ale podtrzymujące palenie, do których zalicza się między innymi : tlen, sprężone powietrze, chlor sprężony, itp. IV grupa – gazy niepalne i nie podtrzymujące palenia, do których zalicza się dwutlenek węgla, gazy szlachetne, dwutlenek siarki , itp.

94
FIZYKOCHEMICZNE WŁASNOŚCI PROCESU PALENIA CIAŁ STAŁYCH

95
Mechanizm spalania się ciał stałych w zależności od ich budowy wewnętrznej
Palność organicznego ciała stałego jest cechą materiału, oznaczającą iż w określonej temperaturze produkty termicznego rozkładu utleniają się aż do wystąpienia spalania. Spalanie materiałów jest poprzedzone ich rozkładem termicznym . Skład i objętość tworzącej się fazy lotnej zależy od struktury materiału i jego właściwości fizycznych oraz od warunków, w których przebiega rozkład termiczny ( między innymi – temperatury rozkładu, szybkości wzrostu temperatury w materiale, zawartości tlenu w otoczeniu, szybkości przepływu powietrza w układzie ). Sposób przekształceń międzyfazowych zależy od rodzaju materiału. Ogólnie ciała stałe pod wpływem ciepła mogą ulegać: rozkładowi, topnieniu, sublimacji, parowaniu, co powoduje , że w początkowym okresie ogrzewania temperatura ciała stałego wzrasta bardzo powoli. Dopiero utlenianie się powstałych produktów lotnych powoduje zmianę stanu energetycznego powstałego układu: ciało stałe – faza lotna i szybki wzrost temperatury ciała.
. Sposób przekształceń międzyfazowych zależy od rodzaju materiału. Ogólnie ciała stałe pod wpływem ciepła mogą ulegać: rozkładowi, topnieniu, sublimacji, parowaniu, co powoduje , że w początkowym okresie ogrzewania temperatura ciała stałego wzrasta bardzo powoli. Dopiero utlenianie się powstałych produktów lotnych powoduje zmianę stanu energetycznego powstałego układu: ciało stałe – faza lotna i szybki wzrost temperatury ciała.")
96
Sposoby tworzenia się fazy lotnej z ciała stałego

97
Zjawiska zachodzące podczas spalania materiałów stałych

98
Zapalenie materiałów stałych
Ad.1 W wyniku ogrzewania ciała stałe ulegają różnorodnym przemianom: mięknięciu, topnieniu lub od razu rozkładowi termicznemu, w wyniku czego tworzy się warstwa węgla. Szybkość ogrzewania się materiału stałego zależy m.in. od pojemności cieplnej materiału tzn. od iloczynu gęstości , współczynnika przewodnictwa cieplnego K i ciepła właściwego cp. Im materiał ma niższą pojemność cieplną tym szybciej ogrzewa się. W tabeli podano przykładowo pojemność cieplną różnych materiałów stałych. .

100
W tabeli podano przykładowo temperatury zapalenia materiałów.
Zapalenie ciała stałego ma miejsce wtedy, gdy powstałe z rozkładu termicznego gazy osiągają stężenie w zakresie ich granic zapalności i temperaturę równą temperaturze zapalenia chociaż jednego składnika. W takich warunkach płomień pojawia się samorzutnie i materiał zaczyna się spalać. Zapalność materiałów stałych zależy od wielu zmiennych, ilości tlenu w otoczeniu, temperatury i fizykochemicznych własności materiałów. Reakcja spalania gazów pirolitycznych, powstałych z rozkładu termicznego jest reakcją egzotermiczną. Jeśli w czasie spalania wydzieli się odpowiednia dla zapewnienia ciągłości spalania energia, płomień zaczyna rozprzestrzeniać się po powierzchni materiału. Ad.3 W tabeli podano przykładowo temperatury zapalenia materiałów.

101
Spalanie tworzyw termoplastycznych
Mechanizm spalania tworzyw termoplastycznych i termoutwardzalnych zależy od budowy tych tworzyw. W czasie rozkładu tworzywa termoplastyczne wytwarzają mieszaninę gazową w wyniku przemian fizycznych (parowanie) oraz chemicznych (rozkład). Powstała faza lotna może ulec zapaleniu lub zapłonowi i w rezultacie spalać się płomieniowo. Mechanizm spalania tych tworzyw można porównać z mechanizmem spalania cieczy. Tworzywo termoutwardzalne rozkłada się pod wpływem ciepła, z wytworzeniem związków niskocząsteczkowych o różnych stanach skupienia. W chwili zapalenia (zapłonu), palna faza lotna zapala się płomieniem, a pozostałość popirolityczna - koksowa zaczyna spalać się bezpłomieniowo czyli tlić na granicy rozdziału fazy stałej i lotnej (powietrza).
 oraz chemicznych (rozkład). Powstała faza lotna może ulec zapaleniu lub zapłonowi i w rezultacie spalać się płomieniowo. Mechanizm spalania tych tworzyw można porównać z mechanizmem spalania cieczy. Tworzywo termoutwardzalne rozkłada się pod wpływem ciepła, z wytworzeniem związków niskocząsteczkowych o różnych stanach skupienia. W chwili zapalenia (zapłonu), palna faza lotna zapala się płomieniem, a pozostałość popirolityczna - koksowa zaczyna spalać się bezpłomieniowo czyli tlić na granicy rozdziału fazy stałej i lotnej (powietrza).")
102
Schemat przepływu ciepła przy spalaniu ciał stałych

104
Chemiczna budowa drewna
Podstawowe składniki drewna. W drewnie występuje około: - 50% węgla, - 42 % tlenu, - 6,1 % wodoru, - 0,04% - 0,26% azotu, - 0, ,2 0% składników mineralnych. Pierwsze miejsce w chemicznej budowie drewna zajmuje celuloza i hemicelulozy , dalsze miejsce zajmują kwasy poliuronowe, mające budowę i charakter polisacharydów ale właściwości kwasowe ( zawierają grupy karboksylowe – COOH); należą tu związki pektynowe i gumy drzewne. Ważnym składnikiem drewna jest lignina, substancja o charakterze aromatycznym, zawierająca grupy metoksylowe – OCH3 i wodorotlenowe –OH. Pod względem funkcjonalnym w drewnie można wyróżnić: - substancję szkieletową, zbudowaną głównie z celulozy, która nadaje jej dużą wytrzymałość , zwłaszcza na rozciąganie; w skład substancji szkieletowej wchodzą również hemicelulozy, - lepiszcze, reprezentowane głównie przez ligninę; poza ligniną w skład lepiszcza wchodzą substancje pektynowe, gumy drzewne i substancje śluzowate, - substancje towarzyszące, a więc żywice, woski, tłuszcze, barwniki, garbniki, alkaloidy, lateksy i inne mniej ważne substancje.
; należą tu związki pektynowe i gumy drzewne. Ważnym składnikiem drewna jest lignina, substancja o charakterze aromatycznym, zawierająca grupy metoksylowe – OCH3 i wodorotlenowe –OH. Pod względem funkcjonalnym w drewnie można wyróżnić: - substancję szkieletową, zbudowaną głównie z celulozy, która nadaje jej dużą. wytrzymałość , zwłaszcza na rozciąganie; w skład substancji szkieletowej wchodzą. również hemicelulozy, - lepiszcze, reprezentowane głównie przez ligninę; poza ligniną w skład. lepiszcza wchodzą substancje pektynowe, gumy drzewne i substancje śluzowate, - substancje towarzyszące, a więc żywice, woski, tłuszcze, barwniki, garbniki, alkaloidy, lateksy i inne mniej ważne substancje.")
105
Obok substancji organicznych w drewnie występują także substancje mineralne, które po spaleniu drewna dają popiół. Udział związków mineralnych w stosunku do ciężaru drewna w stanie powietrznosuchym jest nieznaczny; np. w drewnie sosnowym udział ważniejszych związków mineralnych przedstawia się następująco: Około % popiołu składa się z substancji rozpuszczalnych w wodzie. Są to przede wszystkim węglan potasowy (potaż) K2CO3 i węglan sodowy (soda). Jedna ze stosowanych dawniej, prymitywnych form chemicznego przerobu polegała na wypalaniu drewna na popiół, z którego poprzez ługowanie wodą pozyskiwano potrzebny dla przemysłu potaż. W skład nierozpuszczalnej części popiołu wchodzi przede wszystkim węglan wapnia oraz magnezowe , żelazowe i manganowe sole kwasu węglowego, fosforowego i krzemowego.
 K2CO3 i węglan sodowy (soda). Jedna. ze stosowanych dawniej, prymitywnych form chemicznego przerobu polegała na wypalaniu drewna na popiół, z którego poprzez ługowanie wodą pozyskiwano potrzebny dla przemysłu potaż. W skład nierozpuszczalnej części popiołu wchodzi przede wszystkim węglan wapnia oraz magnezowe , żelazowe i manganowe sole kwasu węglowego, fosforowego i krzemowego.")
106
Celuloza Drewno zbudowane jest w 40 – 60 % z celulozy. W młodych tkankach celulozowe błony komórkowe sklejone są związkami pektynowymi, które tworzą silnie pęczniejącą warstwę międzykomórkową. W dojrzałych tkankach drzewnych substancję sklejającą stanowi lignina. Celuloza występuje w drewnie w mechanicznym i chemicznym powiązaniu z ligniną (wypełniacz) i z hemicelulozami. Celuloza, hemiceluloza i lignina stanowią około 96 % suchej masy drewna. Celulozę o niezmienionych cechach naturalnych, występującą w drewnie drzew żywych , określa się mianem błonnika. Celuloza wyodrębniona z drewna poprzez procesy chemiczne (roztwarzanie drewna) ma skrócone łańcuchy i częściowo zmienione własności. Celuloza jest związkiem wielocząsteczkowym o sumarycznym wzorze Związki wielocząsteczkowe odznaczają się tym , że cząsteczki ich zbudowane są z powtarzających się elementarnych członów, zgrupowanych w długie łańcuchy; związki takie noszą nazwę polimerów.
 i z hemicelulozami. Celuloza, hemiceluloza i lignina stanowią około 96 % suchej masy drewna. Celulozę o niezmienionych cechach naturalnych, występującą w drewnie drzew żywych , określa się mianem błonnika. Celuloza wyodrębniona z drewna poprzez procesy chemiczne (roztwarzanie drewna) ma skrócone łańcuchy i częściowo zmienione własności. Celuloza jest związkiem wielocząsteczkowym o sumarycznym wzorze. Związki wielocząsteczkowe odznaczają się tym , że cząsteczki ich zbudowane są z powtarzających się elementarnych członów, zgrupowanych w długie łańcuchy; związki takie noszą nazwę polimerów.")
107
Podstawowy element, z którego jest zbudowana cząsteczka nosi nazwę monomeru. Stopień polimeryzacji określa liczba powiązanych z sobą monomerów (wartość n ). Łańcuchy celulozy mają różnorodną długość, stąd liczba n przybiera różne wartości. Polimery kondensacyjne odznaczają się tym, że przy łączeniu się monomerów w polimer następuje wydzielanie się wody lub innych substancji. Celuloza jest naturalnym polimerem kondensacyjnym, który powstaje poprzez połączenie się elementarnych cząstek glukozy w długie łańcuchy przy równoczesnym wydzielaniu wody Fizyczne i mechaniczne właściwości oraz techniczna wartość celulozy zależą od długości łańcuchów, czyli od stopnia polimeryzacji. Stopień polimeryzacji (liczba n) określa zmienna liczbę monomerów wchodzących w skład cząsteczki i wynosi dla celulozy od 200 do Wytrzymałość celulozy zwiększa się w miarę wzrostu stopnia polimeryzacji do 700; powyżej tej wartości wytrzymałość wzrasta tylko nieznacznie. Masy celulozowe o stopniu polimeryzacji niższym niż 200 nie nadają się do produkcji materiałów włóknistych.
 określa zmienna liczbę monomerów wchodzących w skład cząsteczki i wynosi dla celulozy od 200 do Wytrzymałość celulozy zwiększa się w miarę wzrostu stopnia polimeryzacji. do 700; powyżej tej wartości wytrzymałość wzrasta tylko nieznacznie. Masy celulozowe o stopniu polimeryzacji niższym niż 200 nie nadają się do produkcji materiałów włóknistych.")
108
Stare papiery i inne materiały celulozowe mają w wyniku starzenia się i związaną z tym depolimeryzacją krótkie łańcuchy ( o stopniu polimeryzacji poniżej 200). Materiały takie są mało wytrzymałe i łatwo się rozpadają w proch (np. wykopaliska egipskie). Celuloza techniczna Celuloza techniczna (masa celulozowa) jest półfabrykatem do produkcji wielu cennych materiałów; najszersze zastosowanie znajduje w produkcji papieru i tkanin. W Polsce podstawowym surowcem do wytwarzania celulozy jest drewno świerkowe, jodłowe i sosnowe, w mniejszych ilościach drewno osikowe i topolowe. Wydajność celulozy zależy od gatunku drewna oraz metody produkcji. Drewno iglaste zawiera więcej celulozy niż drewno liściaste, poza tym długość i smukłość (stosunek długości włókna do jego grubości) włókna celulozowego kształtuje się w drewnie iglastym (zwłaszcza świerkowym) lepiej niż w drewnie liściastym. Wydajność celulozy z różnych gatunków drewna wyrażona w % przedstawia się następująco:
 jest półfabrykatem do produkcji wielu cennych materiałów; najszersze zastosowanie znajduje w produkcji papieru i tkanin. W Polsce podstawowym surowcem do wytwarzania celulozy jest drewno świerkowe, jodłowe i sosnowe, w mniejszych ilościach drewno osikowe i topolowe. Wydajność celulozy zależy od gatunku drewna oraz metody produkcji. Drewno iglaste zawiera więcej celulozy niż drewno liściaste, poza tym długość i smukłość (stosunek długości włókna do jego grubości) włókna celulozowego kształtuje się w drewnie iglastym (zwłaszcza świerkowym) lepiej niż w drewnie liściastym. Wydajność celulozy z różnych gatunków drewna. wyrażona w % przedstawia się następująco:")
109
Hemicelulozy Celulozie towarzyszą węglowodany z grupy polisacharydów, objęte zbiorową nazwą hemiceluloz. Udział hemiceluloz wynosi – zależnie od gatunku drewna od 18 do 35%. Mianem hemiceluloz określa się węglowodany nie rozpuszczające się w wodzie, a ulegające hydrolizie w czasie gotowania z rozcieńczonymi kwasami mineralnymi. Hemicelulozy są to związki niejednolite, obejmujące substancje dwojakiego rodzaju: 1. substancje odporne na działanie kwasów, zbliżone właściwościami do celulozy, 2. substancje ulegające łatwo hydrolizie, zbliżone właściwościami do skrobi. Fizyczne i mechaniczne właściwości hemiceluloz zajmują miejsce pośrednie między właściwościami skrobi i celulozy. W masach celulozowych przeznaczonych do przerobu na papier pożądana jest zawartość hemiceluloz w ilości 6 – 8%. Ich obecność zwiększa zdolność pęcznienia masy, dzięki czemu zwiększa się zdolność spilśniania włókien i zaklejania masy papierniczej. W masach natomiast przeznaczonych do dalszego przerobu chemicznego, zwłaszcza na sztuczne włókno, obecność hemiceluloz jest niepożądana. Mają one inną niż celuloza zdolność reagowania z odczynnikami chemicznymi, co wpływa ujemnie na przebieg i wyniki procesu technologicznego.

110
Przy gotowaniu celulozy część hemiceluloz ulega hydrolizie i w postaci cukrów prostych przechodzi do ługów pocelulozowych. Dzięki temu ług pocelulozowy, powstający przy siarczynowym rozwarstwianiu drewna, można w części wykorzystać do produkcji alkoholu etylowego. W skład substancji ulegających hydrolizie wchodzą głównie pentozany i heksozany. Pentozany występują w drewnie wszystkich gatunków drzew, pierwsze miejsce zajmują w pentozanach ksylan ( C5H8O4)n i araban. Ksylan i araban przechodzą pod wpływem hydrolizy w cukry proste D- ksylozę i L-arabinozę, które nie podlegają fermentacji, natomiast mogą służyć za substrat do produkcji drożdży (białko pastewne). Przy ogrzewaniu z kwasami pentozany wydzielają wodę i przechodzą w furfol C5H4O2, który ma duże znaczenie w przemyśle mas plastycznych. Surowcem do produkcji furfolu są odpady przemysłu drzewnego, np. trociny bukowe lub dębowe , dębowa strużka poekstrakcyjna i inne. Heksozany są to węglowodany występujące w drewnie, o sumaryczny wzorze (C6H10O5)n . Są to polisacharydy o właściwościach mechanicznych zbliżonych do celulozy; najliczniej występuje manan, w znacznie mniejszej ilości galaktan i fruktan. Heksozany ulegają hydrolizie przy ogrzewaniu z rozcieńczonymi kwasami mineralnymi przyłączają wodę, depolimeryzują i przechodzą w heksozy, to jest w cukry proste w myśl wzoru:
n i araban. Ksylan i araban przechodzą pod wpływem hydrolizy w cukry proste D- ksylozę i L-arabinozę, które nie podlegają fermentacji, natomiast mogą służyć za substrat do produkcji drożdży (białko pastewne). Przy ogrzewaniu z kwasami pentozany wydzielają wodę i przechodzą w furfol C5H4O2, który ma duże znaczenie w przemyśle mas plastycznych. Surowcem do produkcji furfolu są odpady przemysłu drzewnego, np. trociny bukowe lub dębowe , dębowa strużka poekstrakcyjna i inne. Heksozany są to węglowodany występujące w drewnie, o sumaryczny wzorze (C6H10O5)n . Są to polisacharydy o właściwościach mechanicznych zbliżonych do celulozy; najliczniej występuje manan, w znacznie mniejszej ilości galaktan i fruktan. Heksozany ulegają hydrolizie przy ogrzewaniu z rozcieńczonymi kwasami mineralnymi przyłączają wodę, depolimeryzują i przechodzą w heksozy, to jest w cukry proste w myśl wzoru: .")
111
Ilościowy udział pentozanów i heksozanów w drewnie poszczególnych gatunków drzew przedstawiono w tabeli. Z liczb zestawionych w tabeli wynika, że w drewnie liściastym udział pentozanów jest 4 –5 razy większy niż heksozanów; w drewnie iglastym pentozany i heksozany występują w ilościach w przybliżeniu równych. Hydroliza drewna, prowadzona w odpowiednio dobranych warunkach powoduje rozkład występującej w drewnie celulozy i hemiceluloz na cukry proste. Cukry te można uważać za produkt końcowy, np. w postaci wykrystalizowanego cukru gronowego, lub traktować je jako produkt pośredni, z którego można otrzymać alkohol etylowy lub drożdże (białko pastewne). Przemysł oparty na hydrolizie drewna rozbudowano do dużych rozmiarów w byłym Związku Radzieckim ; zapoczątkowano go również w Bułgarii. W Polsce i innych krajach Europy hydroliza drewna nie znalazła zastosowania w przemyśle drzewnym. .
. Przemysł oparty na hydrolizie drewna rozbudowano do dużych rozmiarów w byłym. Związku Radzieckim ; zapoczątkowano go również w Bułgarii. W Polsce i innych krajach Europy hydroliza drewna nie znalazła zastosowania w przemyśle drzewnym. .")
112
Substancje pektynowe Substancja międzykomórkowa tworząca blaszki środkowe jest zbudowana w młodych tkankach głównie z substancji pektynowych. W starszych, drewniejących komórkach udział pektyn w blaszkach środkowych maleje, wzrasta natomiast udział ligniny. Przypuszcza się, że w miarę dojrzewania drewna pektyna przechodzi w hemicelulozy, które z kolei przechodzą w ligninę. W skład substancji pektynowych wchodzą kwas galakturonowy, heksozy, i kwasy uronowe. Zawartość pektyny w drewnie iglastym wynosi 4-6% , w drewnie liściastym wynosi 6-8%. Substancje pektynowe zawierają hydrofilowe grupy karboksylowe COOH; wiąże się z tym duża zdolność wchłaniania wody i duże pęcznienie. W produkcji papieru rozpuszczone substancje pektynowe powodują sklejanie się masy papierniczej, co zwiększa wytrzymałość i podwyższa jakość papieru.

113
Lignina Błonnikowy szkielet błony komórkowej jest inkrustowany i otulony ligniną. Lignina jest ciałem bezpostaciowym, o skomplikowanej i niedostatecznie wyjaśnionej budowie chemicznej. Przypisuje się jej wzór chemiczny: W ligninie stwierdzono między innymi około 20% związków aromatycznych oraz grup metoksylowych OCH3 , z których w procesie suchej destylacji drewna powstaje alkohol metylowy. Elementarny skład ligniny ( w porównaniu z celulozą), wyrażony w % suchej substancji przedstawia się następująco:
, wyrażony w % suchej substancji przedstawia się następująco:")
114
Ilościowy udział ligniny w drewnie wynosi od 26 do30 %, zależnie od gatunku drewna i od stopnia zdrewnienia błony komórkowej. Lignina zawiera substancje działające toksycznie na niektóre bakterie i grzyby, wskutek czego jej odporność na procesy rozkładowe jest większa niż odporność czystej celulozy; utrudnia to penetrację bakterii i grzybów, co zwiększa trwałość drewna. W porównaniu z celulozą lignina jest mniej odporna na działanie odczynników chemicznych i rozpuszcza się w alkaliach przy zwiększonym ciśnieniu i podwyższonej temperaturze. W przeciwieństwie do celulozy i hemiceluloz lignina nie ulega hydrolizie pod wpływem kwasów, wobec czego przy scukrzaniu drewna stanowi produkt odpadowy, wymagający dalszego przerobu. Pod wpływem światła lignina żółknie i brunatnieje. Dlatego papiery zawierające ścier drzewny ciemnieją, natomiast papiery wyprodukowane z celulozy bez dodatku ścieru, tzw. Papiery bezdrzewne, nie zmieniają barwy pod wpływem światła. W temperaturze około 130 0C lignina staje się plastyczna, a po ochłodzeniu z powrotem twardnieje . Dzięki temu lignina może stanowić naturalne lepiszcze w produkcji brykietów i płyt trocinowych, płyt pilśniowych i wiórowych oraz materiał wyjściowy do produkcji mas plastycznych.

115
Materiały zapasowe Materiałami zapasowymi w drzewnie są cukry, skrobia i tłuszcze. Wytwarzanie substancji organicznej w liściach drzew odbywa się poprzez fotosyntezę i działanie enzymów. Dzięki energii promieni słonecznych, pochłanianej przez chlorofil w komórkach miękiszowych liści, roślina buduje materię organiczną z wody pobranej z gleby i z dwutlenku węgla pobranego z powietrza, wydzielając przy tym wolny tlen. Jest to reakcja endotermiczna. Produktem asymilacji jest glukoza C6H12O6 . Łatwo rozpuszczalny cukier wędruje w postaci roztworu do komórek miękiszowych łyka, promieni rdzeniowych i miękiszu drzewnego, gdzie zostaje zmagazynowany. Roślina przechowuje materiały zapasowe w postaci skrobi, która ma taki sam wzór sumaryczny jak celuloza; różnica polega na niższym niż u celulozy stopniu polimeryzacji. Skrobia gromadzi się w żywych komórkach bielu w postaci widocznych pod mikroskopem ziaren, zawieszonych w plazmatycznej treści komórek. Materiały zapasowe stanowią podstawę procesów życiowych drzewa; umożliwiają one: wiosenny rozwój pączków i liści, budowę nowo powstającego słoju rocznego, którego znaczna część formuje się przed rozwinięciem liści i rozpoczęciem asymilacji, okresowe wytwarzanie nasion, regulowanie ciśnienia osmotycznego w komórkach przez zmianę skrobi na cukry.

116
Przechowywane materiały pokarmowe występują w postaci związków tłuszczowych i nierozpuszczalnej w wodzie skrobi. Zapasy skrobi mogą być jednak łatwo uruchomione, gdyż skrobia ulega pod wpływem działania fermentu diastazy hydrolizie na cukry, które w postaci roztworu dochodzą do miejsc przeznaczenia. Skrobia gromadzi się w bielu, wypełniając żywe komórki najbardziej zewnętrznych słojów. W miarę zwiększania się zapasów skrobia gromadzi się w głębiej leżących słojach. U buka zasięg skrobi nie przekracza 50 zewnętrznych słojów. W zimie pod wpływem niskich temperatur, skrobia w wewnętrznych słojach drzewa ulega scukrzeniu. Przetworzenie skrobi na rozpuszczalne w wodzie cukry powoduje wzrost stężenia soków w komórkach miękiszowych, wskutek czego obniża się temperatura zamarzania treści komórkowej. W ten sposób zwiększa się odporność komórek a tym samym drzewa, na działanie niskiej temperatury. W przypadku długotrwałych mrozów scukrzenie skrobi posuwa się stopniowo w głąb drzewa, w skrajnych przypadkach cały zasób skrobi ulega scukrzeniu.

117
Wydzieliny drzewne Żywice
W drzewach występują substancje organiczne, które stanowią produkty przemiany materii i nie ulegają dalszym zmianom biochemicznym. Substancje te określone mianem wydzielin, gromadzą się we wnętrzu komórek lub tkanek drzewnych. Do ważnych gospodarczo wydzielin zalicza się żywice, olejki eteryczne, gumy, garbniki, alkaloidy i lateksy. Substancje te wytwarzane są w tkankach i komórkach wydzielniczych; szczególnie ważne spośród nich są przewody żywiczne i rurki mleczne. Żywice W drewnie i w korze drzew iglastych występują związki żywiczne określane mianem żywicy. Żywica odgrywa rolę ochronnego czynnika antyseptycznego ( zabliźnianie uszkodzeń, przeciwdziałanie biologicznemu rozkładowi drewna ), gromadzi się w poziomych i pionowych przewodach żywicznych oraz w komórkach błony komórkowej. W przewodach żywicznych bielu występuje płynna, fizjologicznie czynna żywica, określana mianem żywicy balsamicznej. W toku przemiany bielu na twardziel zwiększa się ilość i zmienia się jakość żywicy. Żywica występująca w twardzieli spełnia rolę wypełniacza oraz środka antyseptycznego, zwiększającego trwałość drewna.
, gromadzi się w poziomych i pionowych przewodach żywicznych oraz w komórkach błony komórkowej. W przewodach żywicznych bielu występuje płynna, fizjologicznie czynna żywica, określana mianem żywicy balsamicznej. W toku przemiany bielu na twardziel zwiększa się ilość i zmienia się jakość żywicy. Żywica występująca w twardzieli spełnia rolę wypełniacza oraz środka antyseptycznego, zwiększającego trwałość drewna.")
118
Żywica balsamiczna jest to roztwór stałych kwasów żywicznych w mieszaninie terpenów (terpentynie). Lotne olejki łatwo ulatniają się w powietrzu, wskutek czego żywica gęstnieje i w końcu krzepnie. Świeżo zebrana żywica jest półpłynna, żółtawa, o barwie i konsystencji świeżego miodu. Brudnoszare zabarwienie żywicy świadczy o dużej zawartości wody i zanieczyszczeniu. Na powietrzu żywica gęstnieje i przybiera kolor biały. Żywica pozyskiwana w toku żywicowania jest zawsze mniej lub więcej zanieczyszczona; średni skład takiej żywicy przedstawia się następująco: Drewno jodły, cisa i jałowca nie zawiera żywicy. Pozostałe krajowe gatunki iglaste zawierają żywicę; najbardziej żywiczne jest drewno sosnowe i modrzewiowe. Słabo żywiczne drewno świerkowe oraz wolne od żywicy drewno jodłowe nadają się dzięki bezwonności do produkcji opakowań ( komplety skrzynkowe, beczki, wełna drzewna ) na produkty spożywcze.
 na produkty spożywcze.")
119
Fizyczna budowa drewna
Określenie drzewo oznacza żyjącą roślinę, natomiast materiał pozyskany ze ściętych drzew jest nazywany drewnem. Swoiste cechy drewna , dostrzegalne gołym okiem, noszą nazwę cech makroskopowych. Natomiast cechy drewna , które można dostrzec tylko pod odpowiednim powiększeniem, nazywają się cechami mikroskopowymi . Cechy te w znacznym stopniu ujawniają techniczne właściwości drewna i są podstawą do rozpoznawania gatunków drewna. Mikroskopowa budowa drewna Do podstawowych elementów budowy mikroskopowej zalicza się: włókna drzewne, naczynia, cewki i komórki miękiszowe. Tworzą one główną masę drewna. Drzewa liściaste mają wszystkie wymienione elementy, drzewa zaś iglaste nie mają włókien drzewnych i naczyń. Na obrazie mikroskopowym widoczne są również wyraźnie promienie rdzeniowe i przewody żywiczne, a między korą a drewnem widać warstwę komórek miazgi.

120
Włókna drzewne Włókna drzewne są to wydłużone, grubościenne, przeważnie martwe komórki wypełnione powietrzem. Pełnią one wyłącznie funkcje mechaniczne. Długość włókien drzewnych wynosi 0,7 - 1,8 mm a średnica 0,02 – 0,05 mm. Gatunki drzew o włóknie wybitnie grubościennym, jak buk, dąb, jesion, mają drewno twarde o dobrych właściwościach mechanicznych. Natomiast topola, lipa, wierzba – o włóknie cienkościennym – mają drewno miękkie o gorszych właściwościach mechanicznych. Włókno dębu

121
Naczynia Naczynia służą do przewodzenia wody wzdłuż pnia. Mają one kształt rur. Ich średnica w drewnie wczesnym niektórych gatunków jest tak duża, że są widoczne gołym okiem na przekrojach – poprzecznym i podłużnym. W drewnie późnym średnica naczyń jest znacznie mniejsza. Średnica naczyń wynosi 0,03 – 0,6 mm, długość do 10cm, a w niektórych gatunkach (dąb) długość naczyń dochodzi do kilku metrów. Naczynia kształtują się w szeregu ułożonych nad sobą członów naczyń , część ścian poprzecznych zanika, pozostałe ściany zostają przebite małymi otworkami; umożliwia to przepływanie przez nie wody w kierunku pionowym. Ruch wody w kierunku promieniowym odbywa się za pośrednictwem jamek występujących w bocznych ściankach naczyń. Naczynia są elementami martwymi, wielokomórkowymi o ścianach zdrewniałych. W czasie formowania się naczyń błona komórki nie grubieje, wskutek czego są to elementy mechanicznie słabe. Przez boczne ścianki naczyń woda przenika do sąsiednich tkanek. Człon naczynia dębu
 długość naczyń dochodzi do kilku metrów. Naczynia kształtują się w szeregu ułożonych nad sobą członów naczyń , część ścian poprzecznych zanika, pozostałe ściany zostają przebite małymi otworkami; umożliwia to przepływanie przez nie wody w kierunku pionowym. Ruch wody w kierunku promieniowym odbywa się za pośrednictwem jamek występujących w bocznych ściankach naczyń. Naczynia są elementami martwymi, wielokomórkowymi o ścianach zdrewniałych. W czasie formowania się naczyń błona komórki nie grubieje, wskutek czego są to elementy mechanicznie słabe. Przez boczne ścianki naczyń woda przenika do sąsiednich tkanek. Człon naczynia dębu.")
122
Cewki Cewki są głównym elementem budowy drewna drzew iglastych . W gatunkach drzew liściastych cewki występują w bardzo małych ilościach. Są to martwe wydłużone komórki o zdrewniałych ściankach różnej grubości. Długość cewek wynosi od 2 – 10 mm, ich grubość jest około 100 razy mniejsza od długości. Najdłuższe są cewki sosny i jodły. Jamki rozmieszczone w bocznych ściankach umożliwiają przenikanie wody z jednej cewki do drugiej. Cewki spełniają w drewnie gatunków iglastych funkcje przewodzenia wody i funkcje mechaniczne. W strefie drewna wczesnego ważniejsze są funkcje przewodzenia wody i dlatego cewki drewna wczesnego są cienkościenne, a ich średnica jest duża. W drewnie późnym ważniejsze są funkcje mechaniczne, dlatego cewki drewna późnego mają grube ścianki, a średnicę 4 – 6 razy mniejszą niż cewki drewna wczesnego. Z tego względu drewno późne gatunków iglastych wykazuje większą gęstość niż drewno wczesne. Cewka w drewnie późnym

123
Komórki miękiszu włóknistego buka
Komórki miękiszowe Komórki miękiszowe są to cienkościenne, żywe komórki o nie zdrewniałych lub słabo zdrewniałych ściankach. W okresie życia drzewa przewodzą one i gromadzą materiały zapasowe lub pełnią funkcje wydzielnicze (np. komórki miękiszowe przewodów żywicznych). Miękisz promieni rdzeniowych i miękisz drzewny, czyli włóknisty, występują u niektórych gatunków drewna liściastego w dużych ilościach, powodując obniżenie ich trwałości. Komórki miękiszu włóknistego buka
. Miękisz promieni rdzeniowych i miękisz drzewny, czyli włóknisty, występują u niektórych gatunków drewna liściastego w dużych ilościach, powodując obniżenie ich trwałości. Komórki miękiszu włóknistego buka.")
124
Przekrój promienia rdzeniowego sosny
Promienie rdzeniowe Promienie rdzeniowe odgrywają rolę łącznika miedzy łykiem a wewnętrznymi warstwami drewna i rdzeniem. Na przekroju poprzecznym przebiegają one w kierunku promieniowym. Składają się z komórek miękiszowych, a u niektórych gatunków (np. sosny) także z poziomo ułożonych cewek przewodzących wodę oraz przewodów żywicznych. Wymiary promieni rdzeniowych są bardzo zróżnicowane. U dębu , wiązu i buka promienie rdzeniowe występują na przekrojach promieniowych w postaci błyszczących smug, zwanych błyszczykiem. W niektórych sortymentach błyszcz jest bardzo ceniony (okleiny deszczułki podłogowe). Przekrój promienia rdzeniowego sosny
 także z poziomo ułożonych cewek przewodzących wodę oraz przewodów żywicznych. Wymiary promieni rdzeniowych są bardzo zróżnicowane. U dębu , wiązu i buka promienie rdzeniowe występują na przekrojach promieniowych w postaci błyszczących smug, zwanych błyszczykiem. W niektórych sortymentach błyszcz jest bardzo ceniony (okleiny deszczułki podłogowe). Przekrój promienia rdzeniowego sosny.")
125
Przewód żywiczny sosny w różnych stadiach wypełniania
Przewody żywiczne Przewody żywiczne W drewnie wielu gatunków drzew iglastych znajdują się przewody żywiczne i żywica (sosna, limba, modrzew, świerk) jodła ma pęcherze żywiczne w korze. Przewody żywiczne bywają podłużne , biegnące wzdłuż pnia i poprzeczne – w promieniach rdzeniowych. Przewody żywiczne mają kształt kanałów wyścielonych komórkami żywicorodnymi. Udział żywicy w normalnym bielu sosny wynosi około 3,95 %, w twardzieli około 5,24 % suchej masy drewna. Występowanie żywicy powoduje zwiększenie trwałości drewna i jest niekiedy zjawiskiem dodatnim. W innych wypadkach (np. w produkcji sklejek, mebli, stolarki budowlanej) jest cechą ujemną, utrudniającą obróbkę. Przewód żywiczny sosny w różnych stadiach wypełniania
 jodła ma pęcherze żywiczne w korze. Przewody żywiczne bywają podłużne , biegnące wzdłuż pnia i poprzeczne – w promieniach rdzeniowych. Przewody żywiczne mają kształt kanałów wyścielonych komórkami żywicorodnymi. Udział żywicy w normalnym bielu sosny wynosi około 3,95 %, w twardzieli około 5,24 % suchej masy drewna. Występowanie żywicy powoduje zwiększenie trwałości drewna i jest niekiedy zjawiskiem dodatnim. W innych wypadkach (np. w produkcji sklejek, mebli, stolarki budowlanej) jest cechą ujemną, utrudniającą obróbkę. Przewód żywiczny sosny w różnych stadiach wypełniania.")
126
Makroskopowa budowa drewna
Miazga Miazga W pniu drzewa między łykiem (wewnętrzną warstwą kory) i drewnem znajduje się warstwa płaskich wzdłużnych cienkościennych komórek, stanowiących tkankę twórczą czyli miazgę. W wyniku podziału miazga odkłada w kierunku rdzenia komórki drewna, na zewnątrz zaś – komórki łyka. Makroskopowa budowa drewna W budowie pnia można odróżnić: rdzeń, drewno, korę (łyko i warstwa korkowa) , promienie rdzeniowe, przewody żywiczne i miazgę. Pierwsze trzy składniki będą omówione w makroskopowej budowie drewna, pozostałe opisane zostały w mikroskopowej budowie drzewa.
 i drewnem znajduje się warstwa płaskich wzdłużnych cienkościennych komórek, stanowiących tkankę twórczą czyli miazgę. W wyniku podziału miazga odkłada w kierunku rdzenia komórki drewna, na zewnątrz zaś – komórki łyka. Makroskopowa budowa drewna. W budowie pnia można odróżnić: rdzeń, drewno, korę (łyko i warstwa korkowa) , promienie rdzeniowe, przewody żywiczne i miazgę. Pierwsze trzy składniki będą omówione w makroskopowej budowie drewna, pozostałe opisane zostały w mikroskopowej budowie drzewa.")
127
Schemat budowy pnia Sosna czteroletnia Dąb czteroletni
1 - łyko, 2 – miazga, 3 – przewód żywiczny, 4 – promienie rdzeniowe (na trzech przekrojach), 5 – granica słoju rocznego, 6 – rdzeń, 7 – warstwa korkowa, 8 – drewno późne, 9 – drewno wczesne, 10 – słój roczny
, 5 – granica słoju rocznego, 6 – rdzeń, 7 – warstwa korkowa, 8 – drewno późne, 9 – drewno wczesne, 10 – słój roczny.")
128
Schemat budowy drewna sosny
1 – słój roczny 2 – promienie rdzeniowe 3 – podłużny przewód żywiczny 4 – drewno wczesne 5 – drewno późne 6 – jamki lejkowate

129
Schemat budowy drewna brzozy
1 – słój roczny 2 – naczynia 3 – promień rdzeniowy 4 – włókna drzewne

130
Schemat budowy drewna dębu
1 – słój roczny 2 – naczynie duże w drewnie wczesnym 3 – naczynie małe w drewnie późnym 4 – wąski promień rdzeniowy 5 – szeroki promień rdzeniowy 6 – włókna drzewne

131
Rdzeń Rdzeń jest fizjologicznym środkiem pnia. Oś rdzenia często jest odchylona od osi geometrycznej pnia. Odchylenie to uwidacznia się na przekroju poprzecznym pnia jako nierównomierna struktura drewna: z jednej strony drewno ma słoje szersze , z drugiej zaś węższe. Na przekroju poprzecznym rdzeń ma zarys okrągły lub owalny, a w niektórych gatunkach drzew – trójkątny (olcha), czworokątny (jesion) lub gwiaździsty (dąb). Średnica rdzenia wynosi 1-5 mm. Na przekroju podłużnym rdzeń ma kształt wąskiego, ciemno zabarwionego paska. Struktura rdzenia jest słaba, gdyż jest on zbudowany ze słabych cienkościennych komórek, które w miarę rozwoju drzewa obumierają. Dlatego rdzeń nie przedstawia wartości technicznej. Drewno Drewno wypełnia przestrzeń między rdzeniem w środku pnia drzewa a warstwą kory na jego obwodzie. Stanowi ono największą część objętości pnia. Drewno ma budowę nierównomierną, jego wygląd i cechy zmieniają się w zależności od kierunku włókien. Jest to materiał typowo różnokierunkowy, co utrudnia obróbkę i zastosowanie techniczne Rozróżnia się w drewnie trzy zasadnicze przekroje:
, czworokątny (jesion) lub gwiaździsty (dąb). Średnica rdzenia wynosi 1-5 mm. Na przekroju podłużnym rdzeń ma kształt wąskiego, ciemno zabarwionego paska. Struktura rdzenia jest słaba, gdyż jest on zbudowany ze słabych cienkościennych komórek, które w miarę rozwoju drzewa obumierają. Dlatego rdzeń nie przedstawia wartości technicznej. Drewno. Drewno wypełnia przestrzeń między rdzeniem w środku pnia drzewa a warstwą kory na jego obwodzie. Stanowi ono największą część objętości pnia. Drewno ma budowę nierównomierną, jego wygląd i cechy zmieniają się w zależności od kierunku włókien. Jest to materiał typowo różnokierunkowy, co utrudnia obróbkę i zastosowanie techniczne Rozróżnia się w drewnie trzy zasadnicze przekroje:")
132
Rodzaje przekrojów drewna
2 1 – przekrój poprzeczny 2 – przekrój podłużny promieniowy 3 – przekrój podłużny styczny 1 3

133
Przekrój poprzeczny 1 Na przekroju poprzecznym są widoczne słoje roczne, ułożone współśrodkowo dookoła rdzenia.

134
Przekrój podłużny promieniowy
2 Na przekroju podłużnym promieniowym przechodzącym przez rdzeń pnia słoje roczne występują w postaci warstw równoległych do rdzenia. Są one przecięte prostopadle do osi pnia smugami promieni rdzeniowych (niewidocznych na rysunku).
.")
135
Przekrój styczny 3 Na przekroju stycznym słoje roczne występują jako smugo paraboliczne. W miarę zbliżania się do rdzenia przekrój styczny staje się coraz bardziej podobny do przekroju promieniowego.

136
Słój roczny - jest to roczny przyrost, który dzięki podziałowi komórek miazgi nakłada się obwodowo na całym drzewie. Najstarsze słoje roczne otaczają rdzeń, najmłodsze są na obwodzie pnia pod korą. Liczba słojów zmniejsza się od odziomka ku wierzchołkowi. Wiek drzewa można zatem określić na podstawie liczby słojów tylko w dolnej części odziomka. W normalnych warunkach wegetacji w jednym roku wytwarza się jeden słój roczny. Wyrazistość słojów rocznych zależy od warunków klimatycznych i gatunku drzewa. W naszym klimacie, powodującym wyraźne odgraniczenie okresu spoczynku (zimą) i okresu wegetacji, słoje roczne są dość wyraźne, natomiast w klimacie podzwrotnikowym słoje roczne tracą swą wyrazistość. Drzewa iglaste mają słoje bardziej wyraźne niż liściaste. Z drzew liściastych: dąb, jesion, wiąz i grochodrzew wykazują większą wyrazistość słojów natomiast brzoza, lipa, osika, grab znacznie mniejszą. Szerokość słoju rocznego mierzy się na promieniu w przekroju poprzecznym pnia. Szerokość tę można wyrażać w mm albo średnią liczbą słojów na 1 cm promienia przekroju poprzecznego pnia. Szerokość słojów bywa różna, w granicach od ułamka milimetra do 20 mm, i zależna od wielu czynników, jak: gatunek drzewa, klimat, gleba, wysokość terenu, naświetlenie, wpływ otoczenia, ilość opadów atmosferycznych i wiele innych. Ten sam gatunek, np.. dąb i jesion, rosnący w zwarciu ma słoje szerokości 1,5 – 4 mm, a na otwartym terenie przy odpowiednim podłożu może mieć słoje dochodzące do 20 mm.
 i okresu wegetacji, słoje roczne są dość wyraźne, natomiast w klimacie podzwrotnikowym słoje roczne tracą swą wyrazistość. Drzewa iglaste mają słoje bardziej wyraźne niż liściaste. Z drzew liściastych: dąb, jesion, wiąz i grochodrzew wykazują większą wyrazistość słojów natomiast brzoza, lipa, osika, grab znacznie mniejszą. Szerokość słoju rocznego mierzy się na promieniu w przekroju poprzecznym pnia. Szerokość tę można wyrażać w mm albo średnią liczbą słojów na 1 cm promienia przekroju poprzecznego pnia. Szerokość słojów bywa różna, w granicach od ułamka milimetra do 20 mm, i zależna od wielu czynników, jak: gatunek drzewa, klimat, gleba, wysokość terenu, naświetlenie, wpływ otoczenia, ilość opadów atmosferycznych i wiele innych. Ten sam gatunek, np.. dąb i jesion, rosnący w zwarciu ma słoje szerokości 1,5 – 4 mm, a na otwartym terenie przy odpowiednim podłożu może mieć słoje dochodzące do 20 mm..")
137
Przekroje sosny wąskosłoistej szerokosłoistej
Szerokość słojów zmniejsza się od rdzenia ku obwodowi. Największą szerokość mają słoje w okolicy szyi korzeniowej. W drewnie korzeni i gałęzi słoje roczne są węższe niż w pniu. Drewno iglaste mające średnio powyżej 4 słojów na 1 cm promienia pnia nazywa się wąskosłoistym, natomiast mające mniej niż 4 słoje na 1 cm – szerokosłoistym. Drewno liściaste zalicza się do wąskosłoistych, jeśli liczba słojów przekracza 6, do średniosłoistych, gdy liczba słojów wynosi od 6 – 3 i szerokosłoistych, jeśli liczba ta jest mniejsza niż 3 na 1 cm promienia przekroju poprzecznego pnia.

138
Drzewo wczesne i późne W słoju rocznym rozróżnia się strefę drewna wczesnego – od strony rdzenia i strefę drewna późnego- zwróconą ku obwodowi. Drewno wczesne składa się z elementów cienkościennych i jest jaśniejsze, drewno późne – z elementów grubościennych i jest ciemniejsze, co szczególnie uwydatnia się w drzewach iglastych. Drewno późne ma większą gęstość i jest bardziej wytrzymałe niż drewno wczesne. A zatem im większy jest udział strefy drewna późnego w stosunku do strefy drewna wczesnego w słoju rocznym, tym większa jest gęstość drewna i lepsze jego właściwości mechaniczne. Drewno iglaste wąskosłoiste jest traktowane w praktyce jako drewno twarde, ciężkie i wytrzymałe, szerokosłoiste zaś jako miękkie, lekkie i słabsze: drewno liściaste odwrotnie wąskosłoiste uważane jest za miękkie i słabsze, a szerokosłoiste za twarde i bardziej wytrzymałe. Pogląd ten, dotyczy drewna liściastego, jest słuszny wtedy, gdy w słojach szerokich znajduje się znaczna ilość drewna późnego. Zagadnienie powyższe ma związek z problemami przemysłowymi o dużym znaczeniu praktycznym. W praktyce wyróżnia się twarde i miękkie drewno dębu i jesionu. Do celów stolarskich i na okleiny używa się drewna wąskosłoistego, gdyż jest miękkie (np. na okleiny- słoje szerokości 1-1,5 mm), do celów kołodziejskich, lotniczych, sportowych itp. Stosuje się drewno szerokosłoiste, jako twarde i bardziej wytrzymałe. Prawidłowa ocena jakości drewna nie powinna polegać wyłącznie na szerokości słojów, lecz na dokładnym określeniu udziału drewna późnego w słojach rocznych i tylko na tej podstawie ustala się jego wartość. Pomiary udziału w surowcu drewna wczesnego i późnego wykonuje się za pomocą specjalnych mikroskopów.
, do celów kołodziejskich, lotniczych, sportowych itp. Stosuje się drewno szerokosłoiste, jako twarde i bardziej wytrzymałe. Prawidłowa ocena jakości drewna nie powinna polegać wyłącznie na szerokości słojów, lecz na dokładnym określeniu udziału drewna późnego w słojach rocznych i tylko na tej podstawie ustala się jego wartość. Pomiary udziału w surowcu drewna wczesnego i późnego wykonuje się za pomocą specjalnych mikroskopów.")
139
Biel i twardziel Na przekrojach poprzecznych i podłużnych wielu gatunków drewna można zauważyć ciemniejsze zabarwienie wewnętrznej części, zwanej twardzielą, podczas gdy obwodowa część drewna ma zabarwienie jaśniejsze i nazywa się bielem. Biel Twardziel Biel przewodzi wodę z solami mineralnymi z korzeni do korony i bierze udział w gromadzeniu minerałów odżywczych. W przewodzeniu wody biorą udział głównie najmłodsze – zewnętrzne słoje bielu. Granica między twardzielą i bielem na przekroju poprzecznym ma zarys nieregularny i nie przebiega wzdłuż jednego słoju. Twardziel nie zawiera komórek żywych i nie bierze udziału w przewodzeniu wody oraz w gromadzeniu materiałów odżywczych, spełnia natomiast funkcje mechaniczne.

140
Szerokość bielu bywa różna w różnych gatunkach drzew, np
Szerokość bielu bywa różna w różnych gatunkach drzew, np. w sośnie biel zajmuje do 80 słojów, czyli kilkanaście centymetrów szerokości, w iwie – zaledwie 8 – 10 słojów, a w cisach tylko do 3 słojów rocznych. Drzewa twardzielowe w okresie młodym mają tylko drewno bielaste. Dopiero po przekroczeniu pewnej granicy wieku (np. w sosnach lat) zaczynają zachodzić zmiany, polegające na drewnieniu i obumieraniu komórek miękiszu drzewnego i promieni rdzeniowych od strony rdzenia. Następuje rozkład treści żywych komórek i powstają substancje twardzielowe (związki żywiczne, garbniki, barwniki), które przesycają błony komórkowe i wypełniają komórki obumarłe, tworząc twardziel. Substancje twardzielowe zwiększają twardość drewna twardzielowego, uodparniając je na gnicie, zmniejszając kurczliwość i zwiększając gęstość. Biel natomiast, mając duże zasoby materiałów odżywczych i wody, po ścięciu drzewa łatwo ulegają rozkładowi; dobrym przykładem nietrwałości bielu jest drewno dębu. Obecność twardzieli zależy nie tylko od gatunku drzewa, lecz także od wielu innych czynników, a między innymi od środowiska, w jakim ono rośnie. Na przykład jesion wytwarza twardziel w wieku 60 – 70 lat. Dlatego jesion ogrodowy, nadający się do ścięcia już po 40 – 50 latach przeważnie nie ma jeszcze twardzieli. Natomiast jesion leśny, który staje się użyteczny dopiero w wieku 80 – 100 lat, ma twardziel. Do grupy drzew z wyraźnie występującą zabarwioną twardzielą należą u nas : dąb, orzech włoski , klon, sosna, modrzew. Do innej grupy należą gatunki drzew z twardzielą nie zabarwioną, jak jodła, świerk, lipa i inne. Twardziel tych drzew, zwłaszcza po wyschnięciu, nie różni się swym zabarwieniem od bielu.
 zaczynają zachodzić zmiany, polegające na drewnieniu i obumieraniu komórek miękiszu drzewnego i promieni rdzeniowych od strony rdzenia. Następuje rozkład treści żywych komórek i powstają substancje twardzielowe (związki żywiczne, garbniki, barwniki), które przesycają błony komórkowe i wypełniają komórki obumarłe, tworząc twardziel. Substancje twardzielowe zwiększają twardość drewna twardzielowego, uodparniając je na gnicie, zmniejszając kurczliwość i zwiększając gęstość. Biel natomiast, mając duże zasoby materiałów odżywczych i wody, po ścięciu drzewa łatwo ulegają rozkładowi; dobrym przykładem nietrwałości bielu jest drewno dębu. Obecność twardzieli zależy nie tylko od gatunku drzewa, lecz także od wielu innych czynników, a między innymi od środowiska, w jakim ono rośnie. Na przykład jesion wytwarza twardziel w wieku 60 – 70 lat. Dlatego jesion ogrodowy, nadający się do ścięcia już po 40 – 50 latach przeważnie nie ma jeszcze twardzieli. Natomiast jesion leśny, który staje się użyteczny dopiero w wieku 80 – 100 lat, ma twardziel. Do grupy drzew z wyraźnie występującą zabarwioną twardzielą należą u nas : dąb, orzech włoski , klon, sosna, modrzew. Do innej grupy należą gatunki drzew z twardzielą nie zabarwioną, jak jodła, świerk, lipa i inne. Twardziel tych drzew, zwłaszcza po wyschnięciu, nie różni się swym zabarwieniem od bielu.")
141
Przekroje poprzeczne jodły
Po wyschnięciu Świeżo ścięta Oddzielną grupę stanowią gatunki beztwardzielowe lub bielaste, w których nie ma różnic między drewnem środkowym i obwodowych części pnia. Przewodzenie wody może tu odbywać się całą powierzchnią przekroju ; najbardziej jednak czynne są obwodowe partie bielu. Do tej grupy należą : buk, grab, jawor, brzoza, osika. Niektóre gatunki drzew wytwarzają w pewnych warunkach twardziel fałszywą.

142
Twardziel fałszywa buku
Pojawia się ona w wyniku samoobrony drzewa pod działaniem czynników destrukcyjnych np. grzybów lub pod wpływem zewnętrznych bodźców, np. mrozu. Fałszywa twardziel, np. buku, uwidacznia się szeroką skalą odcieni barw, które mogą ulec zmianom, zwłaszcza pod wpływem zmian wilgotności drewna. Biel i twardziel wykazują odmienne cechy technologiczne. Drewno twardzielowe ma nieco większą gęstość i nieco lepsze niektóre właściwości mechaniczne; duże są różnice w trwałości: na wolnym powietrzu drewno bielu łatwiej ulega rozkładowi. W razie sztucznego utrwalania bielu i twardzieli, np. podczas nasycania (impregnowania) , wyniki są odwrotne. biel nasyca się łatwo i staje się trwały, a twardziel nie przyjmuje impregnatów zupełnie lub przyjmuje w małej ilości, nie mającej istotnego wpływu na trwałość. Biel sosnowy jest poszukiwany przez przemysł lotniczy i sklejkowy. W drewnie dębowym biel odrzuca się całkowicie. Doświadczenia wykazały, że właściwości techniczne bielu i twardzieli jesionu są praktycznie jednakowe.
 , wyniki są odwrotne. biel nasyca się łatwo i staje się trwały, a twardziel nie przyjmuje impregnatów zupełnie lub przyjmuje w małej ilości, nie mającej istotnego wpływu na trwałość. Biel sosnowy jest poszukiwany przez przemysł lotniczy i sklejkowy. W drewnie dębowym biel odrzuca się całkowicie. Doświadczenia wykazały, że właściwości techniczne bielu i twardzieli jesionu są praktycznie jednakowe.")
143
Właściwości fizyczne drewna
Kora Kora jest złożona z dwóch podstawowych warstw: zewnętrznej tkanki korkowej, stanowiącej osłonę pnia i gałęzi, oraz wewnętrznej – łyka, spełniającej funkcję przewodzenia asymilantów z korony do pnia. W wyniku wzrostu grubości pnia tkanka korkowa pęka i przekształca się w martwą korowinę która stopniowo łuszczy się i odpada warstwami. Nowe warstwy korka wytwarzane są w miarę odpadania korowiny przez tkankę korkotwórczą. Kora zawiera w mniejszych lub większych ilościach garbniki. Pozyskuje się je głównie z kory świerkowej, dębowej i wierzbowej. Kora niektórych drzew, przede wszystkim dębu korkowego, służy jako surowiec do produkcji materiałów izolacyjnych. Właściwości fizyczne drewna Cechy drewna , które można poznać bez naruszenia jego całości i bez zmiany składu chemicznego , noszą nazwę właściwości fizycznych drewna. Do właściwości fizycznych drewna zalicza się: właściwości określające wygląd drewna - barwa, połysk, rysunek, zapach drewna; właściwości wynikające z zachowania się drewna w stosunku do wody – wilgotność, higroskopijność, pęcznienie i kurczenie się drewna; gęstość drewna, właściwości cieplne , elektryczne, akustyczne i inne.

144
Wilgotność drewna W drzewie żyjącym woda stanowi główny składnik soku komórkowego żywych komórek, znajduje się w błonach (ściankach) komórkowych oraz wypełnia cewki i naczynia bielu. W drewnie świeżo ściętym lub mokrym wyróżnia się wodę: a. wodę wolną, wypełniającą cewki i naczynia, b. wodę związaną (higroskopijną), która nasyca błony komórkowe, c. wodę konstytucyjną, stanowiącą część składową związków chemicznych występujących w drewnie, wody tej nie da się usunąć metodami fizycznymi (np. przez suszenie drewna). Najwięcej wody zawierają zewnętrzne warstwy bielu . W drewnie składowanym w wodzie lub spławianym ilość wody wolnej jest większa niż w drewnie świeżo ściętym, natomiast w drewnie powietrzno – suchym wody wolnej nie ma. Wilgotność bezwzględna drewna jest to stosunek masy wody zawartej w drewnie do masy drewna całkowicie suchego. Stosunek ten wyraża się wzorem: W którym W – wilgotność bezwzględna drewna m1 - masa drewna wilgotnego w gramach m2 - masa drewna całkowicie suchego
 komórkowych oraz wypełnia cewki i naczynia bielu. W drewnie świeżo ściętym lub mokrym wyróżnia się wodę: a. wodę wolną, wypełniającą cewki i naczynia, b. wodę związaną (higroskopijną), która nasyca błony komórkowe, c. wodę konstytucyjną, stanowiącą część składową związków chemicznych. występujących w drewnie, wody tej nie da się usunąć metodami. fizycznymi (np. przez suszenie drewna). Najwięcej wody zawierają zewnętrzne warstwy bielu . W drewnie składowanym w wodzie lub spławianym ilość wody wolnej jest większa niż w drewnie świeżo ściętym, natomiast w drewnie powietrzno – suchym wody wolnej nie ma. Wilgotność bezwzględna drewna jest to stosunek masy wody zawartej w drewnie do masy drewna całkowicie suchego. Stosunek ten wyraża się wzorem: W którym W – wilgotność bezwzględna drewna. m1 - masa drewna wilgotnego w gramach. m2 - masa drewna całkowicie suchego.")
145
Stopień wilgotności drewna
Pod wpływem warunków zewnętrznych wilgotność drewna ulega zmianom. W związku z tym wyróżnia się drewno mokre, drewno załadowczo-suche, drewno powierzchnio-suche i drewno suche. Mianem drewna mokrego określa się drewno zawierające wodę wolną, a więc drewno o wilgotności przekraczającej punkt nasycenia włókien. Wilgotność drewna świeżo ściętego waha się , zależnie od gatunku drewna, między 50% a 150% wilgotności. Drewno składowane w wodzie ma wilgotność wyższą niż drewno świeżo ścięte. Mianem drewna załadowczo-suchego określa się drewno o wilgotności 25% lub niższej. Drewno mokre traci pod wpływem składowania na powietrzu część wilgoci i dochodzi do stanu powietrzno-suchego , który w naszych warunkach wyraża się wilgotnością 13 – 22% zależnie od pory roku i wilgotności względnej otaczającego powietrza. Jako wartość przeciętną dla stanu powietrzno-suchego przyjmuje się wilgotność 15%, w krajach zachodnich stan powietrzno-suchy określa się wilgotnością 12%. W naszych warunkach, przy składowaniu drewna na powietrzu nie można osiągnąć wilgotności niższej od stanu powietrzno-suchego, a więc niższej od 13%; dalsze obniżanie wilgotności osiąga się przez suszenie sztuczne.

146
Drewno powietrzno-suche, umieszczone w zamkniętych i zimą ogrzanych pomieszczeniach, traci w dalszym ciągu część swej wody; jego wilgotność ustala się, według danych z literatury, w granicach 8 – 13%. W istocie rzeczy w pomieszczeniach ogrzewanych centralnie wilgotność drewna spada z końcem zimy (marzec) do poziomu 4%, w jesieni wynosi około 13%. Ponadto wyróżnia się wilgotność techniczną, której poziom zależy od wymagań związanych z obróbką drewna, oraz wilgotność użytkową, której poziom zależy od zastosowania i warunków użytkowania drewna lub wyrobów z drewna. Stosowana w przemyśle drzewnym wilgotność techniczna powinna być nieco niższa (około 2%) od wilgotności wymaganej od wyrobów z drewna; w przeciwnym razie przy wysychaniu może nastąpić rozluźnienie połączeń. Zagadnienia wilgotności drewna w różnych zastosowaniach reguluje norma PN-53/D „Stopień wilgotności sortymentów i wyrobów drzewnych”. W myśl postanowień normy wymagana wilgotność użytkowa wynosi:
 do poziomu 4%, w jesieni wynosi około 13%. Ponadto wyróżnia się wilgotność techniczną, której poziom zależy od wymagań związanych z obróbką drewna, oraz wilgotność użytkową, której poziom zależy od zastosowania i warunków użytkowania drewna lub wyrobów z drewna. Stosowana w przemyśle drzewnym wilgotność techniczna powinna być nieco niższa (około 2%) od wilgotności wymaganej od wyrobów z drewna; w przeciwnym razie przy wysychaniu może nastąpić rozluźnienie połączeń. Zagadnienia wilgotności drewna w różnych zastosowaniach reguluje norma PN-53/D „Stopień wilgotności sortymentów i wyrobów drzewnych . W myśl postanowień normy wymagana wilgotność użytkowa wynosi:.")
147
Wilgotności sortymentów i wyrobów drzewnych

148
W myśl postanowień normy pomiar wilgotności wykonuje się przy pomocy wilgotnościomierzy elektrycznych. Wynikająca stąd dokładność pomiaru wynosi (+/-)1,5% wilgotności. Dla uzupełnienia danych zawartych w normie należy dodać , że w konstrukcjach drewnianych nie zamkniętych, lecz osłoniętych dachem stosuje się drewno o wilgotności 15 – 17 %, w konstrukcjach odkrytych o wilgotności 17 – 22%, w budownictwie wodnym drewno o wilgotności 22 – 30%. Stosownie drewna o zbyt dużej wilgotności jest szkodliwe, gdyż powoduje rozluźnienie połączeń, pękanie, paczenie się i nieszczelności w stolarce budowlanej i meblowej, odkształcenia wyrobów drewnianych, zsychanie i rozluźnianie się podłóg oraz rozwój grzyba domowego w budynkach. Nadmierne wysuszenie drewna jest również szkodliwe, gdyż drewno takie pęcznieje i ulega deformacjom i spaczeniu. Charakterystycznym przykładem jest fałdowanie podług wykonanych z deszczułek nadmiernie wysuszonych lub ułożonych na nie wyschniętym podłożu. Racjonalna gospodarka wymaga używania drewna o wilgotności dostosowanej do warunków otoczenia; błędne jest zarówno stosowanie drewna wilgotnego, jak i nadmiernie wysuszonego.
1,5% wilgotności. Dla uzupełnienia danych zawartych w normie należy dodać , że w konstrukcjach drewnianych nie zamkniętych, lecz osłoniętych dachem stosuje się drewno o wilgotności 15 – 17 %, w konstrukcjach odkrytych o wilgotności 17 – 22%, w budownictwie wodnym drewno o wilgotności 22 – 30%. Stosownie drewna o zbyt dużej wilgotności jest szkodliwe, gdyż powoduje rozluźnienie połączeń, pękanie, paczenie się i nieszczelności w stolarce budowlanej i meblowej, odkształcenia wyrobów drewnianych, zsychanie i rozluźnianie się podłóg oraz rozwój grzyba domowego w budynkach. Nadmierne wysuszenie drewna jest również szkodliwe, gdyż drewno takie pęcznieje i ulega deformacjom i spaczeniu. Charakterystycznym przykładem jest fałdowanie podług wykonanych z deszczułek nadmiernie wysuszonych lub ułożonych na nie wyschniętym podłożu. Racjonalna gospodarka wymaga używania drewna o wilgotności dostosowanej do warunków otoczenia; błędne jest zarówno stosowanie drewna wilgotnego, jak i nadmiernie wysuszonego.")
149
Rozprzestrzenianie się płomienia na powierzchni i szybkość liniowa palenia się drewna
Po zapaleniu się drewna w jednym punkcie, płomień stopniowo rozprzestrzenia się na całą jego powierzchnię. Szybkość rozprzestrzeniania się płomienia jest różna i zależy od położenia powierzchni drewna i kierunku rozprzestrzeniania się płomienia. Na poziomej powierzchni płomień rozprzestrzenia się powoli, gdyż są wtedy bardzo niekorzystne warunki nagrzewania się drewna w procesie palenia. Na palącą się powierzchnię ułożoną poziomo ciepło płomienia przekazywane jest tylko drogą promieniowania. Przy powierzchniach ułożonych skośnie do poziomu lub ułożonych pionowo szybkość rozprzestrzeniania się płomienia jest większa i zależy od kierunku rozprzestrzeniania się. Największa szybkość rozprzestrzeniania się płomienia istnieje przy powierzchniach ustawionych pionowo, kiedy płomień rozprzestrzenia się z dołu do góry. W takim układzie nagrzewanie niepalącej się powierzchni drewna odbywa się drogą promieniowania cieplnego oraz bezpośredniego stykania się z nagrzanymi produktami spalania. Przy takim rozprzestrzenianiu się płomienia pole nagrzewającej się powierzchni jest zawsze większe od pola powierzchni, które już spaliło się w tym czasie. Dlatego drewno zawsze „przygotowane” jest do dalszego palenia. Rozprzestrzenianie się płomienia z góry do dołu przebiega z taką samą szybkością , jak na płaszczyźnie poziomej. Szybkość zwęglania drewna waha się w granicach od 0,5 do 1mm/min.

150
Szybkość spalania i łatwość zapalenia ciał stałych ( na przykładzie drewna ) zależnie od i wielkości powierzchni spalania Czas spalania 60 minut minut minuta Wybuch Nie można Trudno Bardzo łatwo Podpalić zapałką

151
Czynniki wpływające na proces palenia się drewna: stosunek ilości do objętości, twardość, wilgotność. Szybkość spalania drewna zależy głównie od: ciężaru objętościowego drewna, jego wilgotności, stosunku powierzchni do objętości, temperatury środowiska i dopływu powietrza. Zamieszczone poniżej rysunki przedstawiają zmianę szybkości spalania w zależności od struktury drewna, jego wilgotności i kształtu. Zależność szybkości spalania drewnianych desek od ich porzecznego przekroju [ długość deski 100 cm ]

152
Największą szybkość spalania ma deska o wym. 1 x 1 x 100 cm
Największą szybkość spalania ma deska o wym. 1 x 1 x 100 cm. Stosunek powierzchni do objętości w tym przypadku równa się 4,02. Najmniejszą szybkość spalania ma deska o wym. 4 x 4 x 100 cm, gdzie stosunek powierzchni do objętości jest dużo mniejszy, równy 1. Z tego powodu, w praktyce, np. mączka drzewna czy wiór zapalają się od płomienia zapałki i spalają się z dużą szybkością podczas gdy np. deska o wymiarach 1 x 1 x 10 cm w tych samych warunkach nie zapali się. Maksymalna szybkość spalania w zależności od ciężaru objętościowego drewna [ próbki o wymiarach 4 x 20 x 100 mm ]

153
Zapalenie różnych gatunków drewna. Temperatura 3200 C
[ wymiary próbek 9 x 9 x 9 mm ]

154
Wpływ ciepła właściwego na czas zapalenia
Cechy palności różnych gatunków drewna

155
Pojemność cieplna dębu, sosny i pianki poliuretanowej
Struktura drewna decyduje o tym, jak szybko dany rodzaj drewna się grzeje to znaczy jak szybko pochłania i magazynuje ciepło, czyli jaką ma pojemność cieplną. O pojemności cieplnej materiałów stałych wspomniano wcześniej w „ Spalaniu materiałów stałych – przy stadiach zapoczątkowania reakcji spalania materiału stałego” – dla przypomnienia jest to iloczyn gęstości , współczynnika przewodnictwa cieplnego K i ciepła właściwego cp. Im materiał ma niższą pojemność cieplną tym szybciej ogrzewa się. W tabeli podano pojemność cieplną dębu , sosny i dla porównania - pianki poliuretanowej. Pojemność cieplna dębu, sosny i pianki poliuretanowej

156
Makrofotopgrafie drewna lipy
Strukturę drewna w tym przypadku lipy i dębu przedstawiają makrofotografie zamieszczone poniżej. Makrofotopgrafie drewna lipy Drewno lekkie – zawiera dużo przestrzeni powietrznych - struktura porowata

157
Makrofotografie drewna dębu
Drewno ciężkie - brak przestrzeni powietrznych – struktura zwarta

158
Podczas ogrzewania tych dwóch gatunków drewna lipa zatrzymuje znacznie więcej ciepła od dębu, ponieważ powietrze zawarte w porach akumuluje ciepło (jest więc izolatorem) podczas gdy części stałe dębu jako lepszy od powietrza przewodnik ciepła odprowadza je szybciej do otoczenia. Dlatego drewno lipy ulega znacznie szybciej zapaleniu w porównaniu z dębem. Gdy porównamy pojemność cieplną, która dla drewna lipy wynosi 1,4 [W2 x s/m4 x K2] natomiast dla drewna dębu [W2 x s/m4 x K2] to aby ogrzać próbkę drewna lipy i dębu do tej samej temperatury, w przypadku drewna lipy potrzeba w przybliżeniu około 2,3 razy mniej ciepła niż w przypadku dębu. Struktury budowy drewna różnych gatunków przedstawiono w temacie „Fizyczna budowa drewna” – w celu porównania ich struktur rysunki drewna dębu, brzozy i sosny przedstawiono w dalszej części opracowania.
![Podczas ogrzewania tych dwóch gatunków drewna lipa zatrzymuje znacznie więcej ciepła od dębu, ponieważ powietrze zawarte w porach akumuluje ciepło (jest więc izolatorem) podczas gdy części stałe dębu jako lepszy od powietrza przewodnik ciepła odprowadza je szybciej do otoczenia. Dlatego drewno lipy ulega znacznie szybciej zapaleniu w porównaniu z dębem. Gdy porównamy pojemność cieplną, która dla drewna lipy wynosi 1,4 [W2 x s/m4 x K2] natomiast dla drewna dębu 3.2 [W2 x s/m4 x K2] to aby ogrzać próbkę drewna lipy i dębu do tej samej temperatury, w przypadku drewna lipy potrzeba w przybliżeniu około 2,3 razy mniej ciepła niż w przypadku dębu.](http://slideplayer.pl/slide/10184559/33/images/158/Podczas+ogrzewania+tych+dw%C3%B3ch+gatunk%C3%B3w+drewna+lipa+zatrzymuje+znacznie+wi%C4%99cej+ciep%C5%82a+od+d%C4%99bu%2C+poniewa%C5%BC+powietrze+zawarte+w+porach+akumuluje+ciep%C5%82o+%28jest+wi%C4%99c+izolatorem%29+podczas+gdy+cz%C4%99%C5%9Bci+sta%C5%82e+d%C4%99bu+jako+lepszy+od+powietrza+przewodnik+ciep%C5%82a+odprowadza+je+szybciej+do+otoczenia.+Dlatego+drewno+lipy+ulega+znacznie+szybciej+zapaleniu+w+por%C3%B3wnaniu+z+d%C4%99bem.+Gdy+por%C3%B3wnamy+pojemno%C5%9B%C4%87+ciepln%C4%85%2C+kt%C3%B3ra+dla+drewna+lipy+wynosi+1%2C4+%5BW2+x+s%2Fm4+x+K2%5D+natomiast+dla+drewna+d%C4%99bu+3.2+%5BW2+x+s%2Fm4+x+K2%5D+to+aby+ogrza%C4%87+pr%C3%B3bk%C4%99+drewna+lipy+i+d%C4%99bu+do+tej+samej+temperatury%2C+w+przypadku+drewna+lipy+potrzeba+w+przybli%C5%BCeniu+oko%C5%82o+2%2C3+razy+mniej+ciep%C5%82a+ni%C5%BC+w+przypadku+d%C4%99bu..jpg "Struktury budowy drewna różnych gatunków przedstawiono w temacie „Fizyczna budowa drewna – w celu porównania ich struktur rysunki drewna dębu, brzozy i sosny przedstawiono w dalszej części opracowania.")
159
8 – drewno późne, 9 – drewno wczesne, 10 – słój roczny
Sosna czteroletnia Dąb czteroletni 1 - łyko, 2 – miazga, 3 – przewód żywiczny, 4 – promienie rdzeniowe (na trzech przekrojach), 5 – granica słoju rocznego, 6 – rdzeń, 7 – warstwa korkowa, 8 – drewno późne, 9 – drewno wczesne, 10 – słój roczny
, 5 – granica słoju rocznego, 6 – rdzeń, 7 – warstwa korkowa, 8 – drewno późne, 9 – drewno wczesne, 10 – słój roczny.")
160
2 – naczynie duże w drewnie wczesnym 3 – naczynie małe w drewnie
1 – słój roczny 2 – naczynie duże w drewnie wczesnym 3 – naczynie małe w drewnie późnym 4 – wąski promień rdzeniowy 5 – szeroki promień rdzeniowy 6 – włókna drzewne Schemat budowy drewna dębu

161
1 – słój roczny 2 – naczynia 3 – promień rdzeniowy 4 – włókna drzewne Schemat budowy drewna brzozy

162
1 – słój roczny 2 – promienie rdzeniowe 3 – podłużny przewód żywiczny 4 – drewno wczesne 5 – drewno późne 6 – jamki lejkowate Schemat budowy drewna sosny

163
Porównując przedstawione struktury budowy drewna, możemy na ich podstawie wysnuć bez odwoływania się do tabel - wnioski odnośnie ich ciężaru objętościowego, współczynnika przewodnictwa cieplnego jak też pojemności cieplnej, a tym samym szybkości zwęglania się. Struktura drewna dębu jest zwarta, nie zawiera powierzchni powietrznych, natomiast w strukturze drewna brzozy takie powierzchnie zaczynają już występować. W strukturze budowy drewna sosny widzimy bardzo dużą ilość takich powierzchni powietrznych. Zwarta struktura drewna dębu ma większą gęstość (stosunek masy do objętości), większa przewodność cieplną – zatem też i większą pojemność cieplną w porównaniu z drewnem brzozy i sosny. Najmniejszą szybkość zwęglania w porównywanych gatunkach drewna będzie miało drewno dębu, największą drewno sosny. Powyższe stwierdzenia potwierdzają temperatury zapalenia ( znajdujące się w tabeli „Cechy palności różnych gatunków drewna” ) i tak temperatura zapalenia dla drewna dębu wynosi 4500C, dla drewna brzozy 3800C , dla drewna sosny 3600C. Różnica między temperaturą zapalenia drewna dębu i temperaturą zapalenia drewna sosny jest bardzo duża bo aż 900C.
, większa przewodność cieplną – zatem też i większą pojemność cieplną w porównaniu z drewnem brzozy i sosny. Najmniejszą szybkość zwęglania w porównywanych gatunkach drewna będzie miało drewno dębu, największą drewno sosny. Powyższe stwierdzenia potwierdzają temperatury zapalenia ( znajdujące się w tabeli „Cechy palności różnych gatunków drewna ) i tak temperatura zapalenia dla drewna dębu wynosi 4500C, dla drewna brzozy 3800C , dla drewna sosny 3600C. Różnica między temperaturą zapalenia drewna dębu i temperaturą zapalenia drewna sosny jest bardzo duża bo aż 900C.")
164
Struktura drewna zmienia się wraz z jego wiekiem i zależna jest od tego czy dany gatunek drzewa utworzył już w swej strukturze twardziel czy nie i czy taką twardziel dany gatunek drzewa tworzy. Inne własności będzie miało drewno, które w ciągu swojego wzrostu nie utworzyło jeszcze twardzieli i inne własności będzie miało to samo drewno zawierające twardziel. Twardość drewna tego samego gatunku w tym przypadku może być różna, zatem i pojemność cieplna tego samego drewna może być różna – jego ciężar objętościowy, ciepło właściwe jak i przewodność cieplna. Drewno tego samego gatunku pozyskane w różnym okresie swojego wzrostu zakładając, że jego wilgotność jest taka sama , może zapalać się w różniących się od siebie temperaturach.

165
Wpływ wilgotności drewna na jego proces palenia
Wilgotność drewna ma istotny wpływ na jego szybkość palenia się. Ten sam gatunek drewna o różnej wilgotności, będzie się spalał z inną szybkością. Pojemność cieplna drewna suchego ( o mniejszym % wilgotności) będzie mniejsza od pojemności cieplnej tego samego drewna lecz o większej wilgotności. Aby ogrzać do tej samej temperatury drewno o większej wilgotności musimy dostarczyć więcej energii cieplnej. Część energii cieplnej (egzotermicznej ) uzyskana w wyniku spalania i zwrócona do materiału będzie pomniejszona o energię niezbędną do odparowania wody w drewnie o większej [ % ] wilgotności. Szybkość spalania materiałów stałych w płomieniu dyfuzyjnym wyrażona jest wzorem: m - szybkość spalania [g/m2 x s], QF - strumień ciepła dostarczony z płomienia do powierzchni [KW/m2], QL - strumień ciepła tracony z powierzchni w wyniku promieniowania powiększony o ciepło potrzebne na odparowanie wilgoci [kW/m2] LV - jest to ciepło gazyfikacji tzn. ilość ciepła [kJ], niezbędna do powstania 1 grama substancji lotnych [kJ/g]
 będzie mniejsza od pojemności cieplnej tego samego drewna lecz o większej wilgotności. Aby ogrzać do tej samej temperatury drewno o większej wilgotności musimy dostarczyć więcej energii cieplnej. Część energii cieplnej (egzotermicznej ) uzyskana w wyniku spalania i zwrócona do materiału będzie pomniejszona o energię niezbędną do odparowania wody w drewnie o większej [ % ] wilgotności. Szybkość spalania materiałów stałych w płomieniu dyfuzyjnym wyrażona jest wzorem: m - szybkość spalania [g/m2 x s], QF - strumień ciepła dostarczony z płomienia do powierzchni [KW/m2], QL - strumień ciepła tracony z powierzchni w wyniku promieniowania. powiększony o ciepło potrzebne na odparowanie wilgoci [kW/m2] LV - jest to ciepło gazyfikacji tzn. ilość ciepła [kJ], niezbędna do. powstania 1 grama substancji lotnych [kJ/g]")
166
Różnica QF – QL dla drewna suchego będzie mniejsza niż dla drewna o większej zawartości wilgoci, LV - dla drewna suchego i wilgotnego nie zmienia swojej wartości, w związku z tym (m) dla drewna suchego będzie większa w porównaniu z ( m ) dla drewna o większej wilgotności. Wilgotność drewna spowalnia szybkość jego spalania aż do zaniku procesu palenia

167
Palenie się ciał strzępiastych
Tworzywa i paliwa stałe charakteryzują się małym ciepłem właściwym i złym przewodzeniem ciepła, co przyczynia się do szybkiego nagrzewania ich powierzchni i powolnych przemian warstw położonych głębiej. Jeżeli jednak materiał ma dużą powierzchnię właściwą wówczas o szybkości spalania decyduje głównie nagrzanie powierzchniowe. Dlatego najbardziej pożarowo niebezpieczne są : - materiały włókniste i inne w stanie rozdrobnionym (np. bawełna, wełna drewno, wióry), - materiały spienione i gąbczaste (np. gąbki lateksowe, pianki), - materiały w postaci folii, arkuszy, płyt itp. składowane w stanie zapewniającym obustronny dostęp powietrza Szybkość spalania materiału można obniżyć przez zastosowanie środków ogniochronnych nakładanych na powierzchnię lub wbudowanie w strukturę tworzywa związków/cząsteczek nadających im własność samogaśnięcia lub utrudniających palność (retardantów).
, - materiały spienione i gąbczaste (np. gąbki lateksowe, pianki), - materiały w postaci folii, arkuszy, płyt itp. składowane w stanie. zapewniającym obustronny dostęp powietrza. Szybkość spalania materiału można obniżyć przez zastosowanie środków ogniochronnych nakładanych na powierzchnię lub wbudowanie w strukturę tworzywa związków/cząsteczek nadających im własność samogaśnięcia lub utrudniających palność (retardantów).")
168
Działanie tych środków polega na: ograniczeniu dostępu tlenu oraz izolacji termicznej materiału palnego przez tworzenie spienionej warstwy izolacyjnej, modyfikacji przebiegu pirolizy w kierunku zwiększenia udziału pozostałości stałej i ograniczenia ilości wydzielanych gazów palnych, wpływaniu na przebieg reakcji spalania przez wytwarzanie lotnych inhibitorów- katalizatorów ujemnych, zwalniających reakcją spalania.

169
Obserwacja faz palenia się drewna

170
Podczas ogrzewania drewna zachodzą w nim następujące zmiany: w temperaturze około 110°C zaczyna odparowywać woda oraz zawarte w drewnie olejki eteryczne (pojawiają się więc pierwsze palne produkty ), w temperaturze około 150°C ulatniają się żywice oraz zaadsorbowane gazy oraz niektóre produkty rozkładu (pojawiaj, się więc dalsze palne produkty), - w temperaturze około 230°C zaczyna się pojawiać powierzchniowe brunatnienie (początek zwęglania ), - w temperaturze około 270°C tworzy się pyroforyczny węgiel o zdolnościach do samozapalenia, związane jest to ze zwęglaniem się hemiceluloz, - w wyższych temperaturach zaczyna tworzyć się większa ilość węgla drzewnego związana ze zwęglaniem głównych składników drewna tj.celuloz i lignin Pod działaniem ciepła następuje najpierw rozkład warstw zewnętrznych drewna, wytwarzają się przy tym takie gazy palne jak: tlenek węgla, metan, etylen oraz pojawiają się pary acetonu, metanolu itp. Przy dalszym wzroście temperatury następuje na skutek spalania się gazów rozkład dalszych, głębszych warstw. Drewno zwęgla się, przy czym proces ten zaczyna przebiegać ze znaczną, szybkością już w temperaturze °C. Proces zwęglania się , a raczej jego szybkość nie jest jednakowa dla wszystkich gatunków drewna.

171
Kolejność ta jest następująca (w kierunku malejącym) buk, brzoza, sosna, świerk. Szybkość zwęglania drewna waha się w granicach od 0,5 do 1mm/min. Na skutek przewodnictwa , konwekcji i promieniowania ciepło przenika do coraz głębszych warstw drewna, ale z drugiej strony zaczyna narastać grubość zwęglonej warstwy, która działa izolująco. Dlatego też podczas spalania drewna po pewnym czasie następuje obniżenie temperatury spalania. Przy odpowiedniej grubości drewna po pewnym czasie zaobserwować można ponowny wzrost temperatury spalania drewna, co związane jest z niszczeniem ochronnej warstwy izolującej węgla pod wpływem par i gazów wydobywających się z głębszych warstw. Działanie ochronne powłoki węgla ustaje całkowicie z chwila jej rozżarzenia się. Drewno spala się wówczas bardzo szybko. Istotne znaczenie ma w tym przypadku przekrój drewna. Duża rolę odgrywa też stosunek objętości drewna do jego powierzchni. Niszczenie drewna postępuje z pewną regularnością, i można przewidzieć jego postęp, zniszczoną część można dość ściśle rozgraniczyć od części nienaruszonej, postęp niszczenia drewna o dużych przekrojach jest proporcjonalny do czasu.

172
Określenie temperatury zapalenia
Temperatura zapalenia – jest to najniższa temperatura materiału, który ogrzewany strumieniem ciepła dostarczonym z zewnątrz w wyniku rozkładu termicznego wydziela palną fazę lotną o stężeniu umożliwiającym jego zapalenie się – to znaczy samorzutne pojawienie się płomienia. Zapoczątkowanie reakcji spalania ciała stałego może nastąpić (podobnie jak cieczy i gazów) pod wpływem zewnętrznych źródeł ciepła czyli zapłonu jak i też wskutek dostarczenia strumienia ciepła promieniowania tzn. zapalenia lub samozapalenia. Zjawisko zapłonu ciał stałych można zaobserwować w warunkach ogrzewania powierzchni ciała stałego. Pod wpływem ciepła powstałe w wyniku rozkładu powierzchni ciała stałego produkty pirolizy mogę ulec zapłonowi pomimo iż cały materiał nie jest ogrzewany. Oznacza to, że temperatura zapłonu ciała stałego nie może być definiowana w odniesieniu do całej masy ogrzewanego materiału a jedynie do temperatury jej powierzchni. Tworzenie się bowiem palnych produktów lotnych poprzedzone jest rozkładem termicznym ciała stałego, który jest procesem nieodwracalnym. Nie jest to więc proces równoważny zapłonowi cieczy, który może być obliczony z zależności równowagowych para - ciecz w chwili zapłonu. Dlatego też temperatura zapłonu ciała stałego może być odniesiona tylko do temperatury powierzchni ciała stałego, przy której następuje już zapłon produktów rozkładu od zewnętrznych źródeł ciepła.
 pod wpływem zewnętrznych źródeł ciepła czyli zapłonu jak i też wskutek dostarczenia strumienia ciepła promieniowania tzn. zapalenia lub samozapalenia. Zjawisko zapłonu ciał stałych można zaobserwować w warunkach ogrzewania powierzchni ciała stałego. Pod wpływem ciepła powstałe w wyniku rozkładu powierzchni ciała stałego produkty pirolizy mogę ulec zapłonowi pomimo iż cały materiał nie jest ogrzewany. Oznacza to, że temperatura zapłonu ciała stałego nie może być definiowana w odniesieniu do całej masy ogrzewanego materiału a jedynie do temperatury jej powierzchni. Tworzenie się bowiem palnych produktów lotnych poprzedzone jest rozkładem termicznym ciała stałego, który jest procesem nieodwracalnym. Nie jest to więc proces równoważny zapłonowi cieczy, który może być obliczony z zależności równowagowych para - ciecz w chwili zapłonu. Dlatego też temperatura zapłonu ciała stałego może być odniesiona tylko do temperatury powierzchni ciała stałego, przy której następuje już zapłon produktów rozkładu od zewnętrznych źródeł ciepła.")
173
Ponieważ temperatura powierzchni może być określana na podstawie bilansu cieplnego opisującego warunki zapłonu prowadzące do rozwinięcia się ciągłego spalania powierzchni materiału, istnieją w literaturze przedmiotu równania analityczne określające bilanse na powierzchni ogrzewanych ciał stałych ale ich interpretacja wykracza poza zakres niniejszego opracowania. W Polsce nie istnieje normatywna metoda wyznaczania temperatury zapłonu ciał stałych. Nie stosuje się też obliczeniowych metod wyznaczania tej wielkości.

174
SPALANIE I ROZKŁAD TWORZYW SZTUCZNYCH

175
produkty stałe - zwykle zwęglone pozostałości, węgiel lub popiół,
Spalanie tworzyw sztucznych to proces, który obejmuje wiele przemian fizycznych i chemicznych, w czasie których powstaje mieszanina substancji chemicznych o złożonym składzie. W zależności od rodzaju tworzywa, mogą wydzielać się następujące produkty: gazy palne lub pary, które spalają się w obecności powietrza (metan, etan, etylen, formaldehyd, aceton, tlenek węgla), gazy niepalne lub gazy, które nie palą się w obecności powietrza (dwutlenek węgla, chlorowodór, bromowodór, para wodna), ciecze, zwykle częściowo rozłożony polimer i związki organiczne o wysokim ciężarze cząsteczkowym, produkty stałe - zwykle zwęglone pozostałości, węgiel lub popiół, porwane cząsteczki stałe lub fragmenty polimeru jako dym.
, gazy niepalne lub gazy, które nie palą się w obecności powietrza (dwutlenek węgla, chlorowodór, bromowodór, para wodna), ciecze, zwykle częściowo rozłożony polimer i związki organiczne o wysokim ciężarze cząsteczkowym, produkty stałe - zwykle zwęglone pozostałości, węgiel lub popiół, porwane cząsteczki stałe lub fragmenty polimeru jako dym.")
176
brom, chlor, fosgen (Br2, Cl2, COCl2),
Podczas rozkładu termicznego i spalania polimerów powstaje wiele gazowych związków toksycznych, takich jak: tlenki węgla (CO i CO2), tlenki azotu (NO i NO2), amoniak (NH3), brom, chlor, fosgen (Br2, Cl2, COCl2), cyjanowodór, siarkowodór, dwutlenek siarki (HCN, H2S, SO2), chlorowodór, bromowodór, fluorowodór (HCl, HBr, HF), aldehydy i inne.
, tlenki azotu (NO i NO2), amoniak (NH3), brom, chlor, fosgen (Br2, Cl2, COCl2), cyjanowodór, siarkowodór, dwutlenek siarki (HCN, H2S, SO2), chlorowodór, bromowodór, fluorowodór (HCl, HBr, HF), aldehydy i inne.")
177
Ponieważ polichlorek winylu jest jednym z najczęściej występujących tworzyw sztucznych z którym mogą zetknąć się ratownicy podczas prowadzenia akcji ratowniczej w warunkach pożaru, poniżej przedstawiono jego charakterystykę w rozkładzie pirolitycznym. Polichlorek winylu podczas pirolitycznego rozkładu jest jednym z najbardziej niebezpiecznych polimerów ze wzglądu na tworzenie się chlorowodoru tj.: przy 140°C - następują początki utleniania się chlorowodoru 210°C - początek topnienia się PVC, wydziela się 60 % HC1 300°C - utlenia się 85 % zawartości HC l/kg 400°C - utlenia się 91 % zawartości HC l/kg 530°C - utlenia się 99 % zawartości HCL l/kg pow. 530°C - polichlorek winylu (polwinit) zapala się. Tworzący się chlorowodór oprócz działania inhibicyjnego na płomień, posiada silne właściwości korodujące.
 zapala się. Tworzący się chlorowodór oprócz działania inhibicyjnego na płomień, posiada silne właściwości korodujące.")
178
WSKAŹNIK TLENOWY JAKO PARAMETR PORÓWNAWCZY OCENY ZAPALNOŚCI TWORZYW SZTUCZNYCH
Wskaźnik tlenowy jest to najmniejsza wyrażona w procentach objętościowych zawartość tlenu w mieszaninie tlenu i azotu, która w warunkach metody badań podtrzymuje stałe palenie się badanej próbki. Metodę pomiaru wskaźnika tlenowego stosuje się do wszystkich tworzyw sztucznych w celu porównawczej oceny ich zapalności. Stosuje się ją przy ocenie modyfikacji tego samego rodzaju tworzywa oraz ocenie wpływu dodatku wypełniaczy i plastyfikatorów na spalanie tworzywa i skuteczności działania antypirenów. Wskaźnik tlenowy nie stanowi podstawy do klasyfikacji tworzyw pod względem zagrożenia pożarowego. Opierając się na wartości wskaźnika tlenowego, wprowadzono w przemyśle amerykańskim (wykorzystywany również w Polsce) podział na tzw. tworzywa: palne, gdy wskaźnik tlenowy WT< 21 %, samogasnące, gdy wskaźnik tlenowy 21 % < WT < 28 %, niepalne, gdy wskaźnik tlenowy 28 % < WT < 100 %. Wyznaczanie wartości wskaźnika tlenowego odbywa się metodą laboratoryjną zgodnie z polską normą PN-ISO „Oznaczanie zapalności metodą wskaźnika tlenowego”.
 podział na tzw. tworzywa: palne, gdy wskaźnik tlenowy WT< 21 %, samogasnące, gdy wskaźnik tlenowy 21 % < WT < 28 %, niepalne, gdy wskaźnik tlenowy 28 % < WT < 100 %. Wyznaczanie wartości wskaźnika tlenowego odbywa się metodą laboratoryjną zgodnie z polską normą PN-ISO „Oznaczanie zapalności metodą wskaźnika tlenowego .")
179
Największe zagrożenie dla życia i zdrowia ludzi podczas pożaru tworzyw sztucznych stwarzają poliamid, polichlorek winylu oraz poliuretan, według następującego uszeregowania: poliamid > polichlorek winylu > poliuretan > polistyren > polietylen > polipropylen > polimetakrylan metylu.

180
O CZYM NALEŻY PAMIĘTAC PODZCZAS PROWADZENIA DZIAŁAŃ RATOWNICZYCH
zapach środków toksycznych np. chlorowodoru, chlorku winylu, dymów spalinowych itp. jest wyczuwalny znacznie wcześniej niż stężenie środka toksycznego stanie się groźne dla życia człowieka. w czasie spalania lub pirolizy (rozkładu termicznego) polichlorku winylu powstaje chlorowodór, który doskonale rozpuszcza się w wodzie, tworząc roztwór kwasu solnego. Stężenie kwasu solnego będzie uzależnione od ilości chlorowodoru oraz ilości wody, zwłaszcza użytej do gaszenia. Kwasowość wody odprowadzonej do instalacji ściekowo - kanalizacyjnej podczas ewentualnego pożaru, o wartości poniżej 5 pH wymaga jej neutralizacji (w przypadku obecności chlorowodoru i kwasu solnego papierek lakmusowy zabarwia się na różowo). osoby ratujące powinny wiedzieć, że niektóre gazy nie wywołują zatrucia bezpośredniego po narażeniu na ich działanie. Objawy zatrucia mogą występować po kilku godzinach a nawet po dwóch dobach. Z tego względu wszystkie osoby podejrzane o zatrucie gazami działającymi w ten sposób powinny znaleźć się pod opieką lekarską.
 polichlorku winylu powstaje chlorowodór, który doskonale rozpuszcza się w wodzie, tworząc roztwór kwasu solnego. Stężenie kwasu solnego będzie uzależnione od ilości chlorowodoru oraz ilości wody, zwłaszcza użytej do gaszenia. Kwasowość wody odprowadzonej do instalacji ściekowo - kanalizacyjnej podczas ewentualnego pożaru, o wartości poniżej 5 pH wymaga jej neutralizacji (w przypadku obecności chlorowodoru i kwasu solnego papierek lakmusowy zabarwia się na różowo). osoby ratujące powinny wiedzieć, że niektóre gazy nie wywołują zatrucia bezpośredniego po narażeniu na ich działanie. Objawy zatrucia mogą występować po kilku godzinach a nawet po dwóch dobach. Z tego względu wszystkie osoby podejrzane o zatrucie gazami działającymi w ten sposób powinny znaleźć się pod opieką lekarską.")
181
O CZYM NALEŻY PAMIĘTAĆ PODCZAS PROWADZENIA DZIAŁAŃ RATOWNICZYCH
W przypadku wystąpienia w powietrzu toksycznych stężeń gazu ratujący zabezpieczony odzieżą ochronną i izolującym aparatem oddechowym powinien: usunąć natychmiast zatrutego (wyprowadzić, wynieść na rękach, na plecach lub płachcie) z miejsca w którym jest narażony na działanie gazu, zdjąć z poszkodowanego skażoną odzież, zapewnić dopływ świeżego powietrza, spokój i poczucie bezpieczeństwa, ułożyć zatrutego wygodnie i zapewnić mu ciepło (okryć kocami lub przekładać do nóg ciepłe termofory owinięte w ręczniki), rozluźnić wszystkie uciskające części garderoby, u nieprzytomnego sprawdzić i utrzymywać drożność dróg oddechowych: głowę leżącego skręcić lekko w lewo, odsysać ssakiem lub wygarniać palcem śluz z jamy ustnej i gardła, usunąć protezy zębowe, podtrzymywać szczękę, kontrolować czy nie zapada się język; osobie nieprzytomnej nie wolno podawać żadnych środków doustnie, gdyż w tym stanie nie ma ona odruchu połykania i może dojść do zachłystowego zapalenia płuc lub uduszenia, w przypadku zaburzonego oddechu podawać tlen aż do chwili powrotu prawidłowego oddychania; tlen podaje się przez maskę twarzową lub cewnik nosowy z szybkością 4-6 dm3/min, przy zatrzymaniu oddechu należy wykonać sztuczne oddychanie przy użyciu worka Ambu, a w przypadku jego braku metodą usta-usta; nie należy wykonywać sztucznego oddychania w zatruciach zagrażających obrzękiem płuc; nie wolno wykonywać sztucznego oddychania w przypadku krwotoku z gardła lub gdy może dojść do zatrucia ratującego, w razie ustającej akcji serca wykonywać zabiegi podtrzymujące akcję serca tj. zewnętrzny masaż serca, przygotować zatrutego do transportu, zapewnić zatrutemu jak najszybciej pomoc lekarską.
 z miejsca w którym jest narażony na działanie gazu, zdjąć z poszkodowanego skażoną odzież, zapewnić dopływ świeżego powietrza, spokój i poczucie bezpieczeństwa, ułożyć zatrutego wygodnie i zapewnić mu ciepło (okryć kocami lub przekładać do nóg ciepłe termofory owinięte w ręczniki), rozluźnić wszystkie uciskające części garderoby, u nieprzytomnego sprawdzić i utrzymywać drożność dróg oddechowych: głowę leżącego skręcić lekko w lewo, odsysać ssakiem lub wygarniać palcem śluz z jamy ustnej i gardła, usunąć protezy zębowe, podtrzymywać szczękę, kontrolować czy nie zapada się język; osobie nieprzytomnej nie wolno podawać żadnych środków doustnie, gdyż w tym stanie nie ma ona odruchu połykania i może dojść do zachłystowego zapalenia płuc lub uduszenia, w przypadku zaburzonego oddechu podawać tlen aż do chwili powrotu prawidłowego oddychania; tlen podaje się przez maskę twarzową lub cewnik nosowy z szybkością 4-6 dm3/min, przy zatrzymaniu oddechu należy wykonać sztuczne oddychanie przy użyciu worka Ambu, a w przypadku jego braku metodą usta-usta; nie należy wykonywać sztucznego oddychania w zatruciach zagrażających obrzękiem płuc; nie wolno wykonywać sztucznego oddychania w przypadku krwotoku z gardła lub gdy może dojść do zatrucia ratującego, w razie ustającej akcji serca wykonywać zabiegi podtrzymujące akcję serca tj. zewnętrzny masaż serca, przygotować zatrutego do transportu, zapewnić zatrutemu jak najszybciej pomoc lekarską.")
182
Podstawowe pojęcia z zakresu samonagrzewania i samozapalenia
Samonagrzewanie poprzedza samozapalenie i jest to proces charakteryzujący się tym, że temperatura układu palnego (materiał palny ulegający samonagrzewaniu – powietrze lub inny utleniacz) podwyższa się w wyniku generacji ciepła wewnątrz samego układu niezależnie od dopływu strumienia ciepła z zewnątrz. Jeśli wzrost szybkości wydzielania się ciepła jest bardzo duży, to zjawisko samonagrzewania przyspiesza się z możliwością przejścia w proces spalania (samozapalenia). Temperatura układu palnego, przy której zaczyna się już samoprzyspieszenie reakcji i akumulacja ciepła, nazywa się temperaturą krytyczną samonagrzewania. Czas od momentu ogrzewania się substancji (ciepłem będącym wynikiem reakcji chemicznych, procesów biologicznych, zjawisk fizycznych) do pojawienia się płomienia nazywa się okresem indukcji. Samonagrzewaniu (samozapaleniu) mogą ulegać: gazy, ciecze i ciała stałe. Samonagrzewanie materialów może zachodzić w różny sposób i może być wynikiem : • reakcji chemicznych (utlenianie, polimeryzacja), • zjawisk fizycznych (adsorpcja), • przemian biologicznych (rozmnażanie, oddychanie komórek roślinnych).
 podwyższa się w wyniku generacji ciepła wewnątrz samego układu niezależnie od dopływu strumienia ciepła z zewnątrz. Jeśli wzrost szybkości wydzielania się ciepła jest bardzo duży, to zjawisko samonagrzewania przyspiesza się z możliwością przejścia w proces spalania (samozapalenia). Temperatura układu palnego, przy której zaczyna się już samoprzyspieszenie reakcji i akumulacja ciepła, nazywa się temperaturą krytyczną samonagrzewania. Czas od momentu ogrzewania się substancji (ciepłem będącym wynikiem reakcji chemicznych, procesów biologicznych, zjawisk fizycznych) do pojawienia się płomienia nazywa się okresem indukcji. Samonagrzewaniu (samozapaleniu) mogą ulegać: gazy, ciecze i ciała stałe. Samonagrzewanie materialów może zachodzić w różny sposób i może być wynikiem : • reakcji chemicznych (utlenianie, polimeryzacja), • zjawisk fizycznych (adsorpcja), • przemian biologicznych (rozmnażanie, oddychanie komórek roślinnych).")
183
zjawisko fizyczne spalanie
samonagrzewanie płomieniowe chemiczna reakcja materiału samozapalenie procesy biologiczne tlenie

184
– – – – – – okres indukcji Wykres zmian temperatury w funkcji czasu samonagrzewania się materiału palnego

185
Samonagrzewanie pod wpływem reakcji chemicznych
Wszystkie materiały, które ulegają samonagrzewaniu pod wpływem reakcji chemicznych można podzielić na: • materiały, które ulegają samonagrzewaniu pod wpływem powietrza (oleje roślinne, fosfor biały (żółty), siarczki, węgliki alkalicznych metali, np: N2S). Wszystkie te substancje zdolne są utleniać się na powietrzu z wydzielaniem ciepła, kosztem którego reakcja przyspiesza się i następuje samonagrzewanie i w konsekwencji może nastąpić samozapalenie. Czas indukcji dla tych materiałów jest bardzo różny; • materiały, które ulegają samonagrzewaniu pod wpływem wilgoci; do tej grupy należą: sód, potas, rubid, cez, lit ( pierwiastki I grupy układu okresowego), jak również tlenki metali alkalicznych. Wszystkie te substancje energicznie łączą się z wodą, wydziela się wówczas ciepło, które powoduje wzrost temperatury związku chemicznego i w konsekwencji samonagrzewanie, ewentualnie samozapalenie, np. w reakcji 2Na + 2H2O → 2NaOH + H2 • materiały, które ulegają samonagrzewaniu w mieszaninach między sobą. Mieszaniny samozapalające otrzymuje się w wyniku zmieszania utleniacza z materiałem palnym. Najsilniejsze utleniacze to: kwas HNO3 i jego sole, szczególnie azotany I grupy układu okresowego kwas siarkowy H2SO4 nadtlenki metali I i II grupy układu okresowego, np. N2O2 wszystkie tlenowe kwasy pochodne od chloru, np.KClO2 obok tlenu silnym utleniaczem jest chlor; acetylen, wodór, w mieszaninie z chlorem samozapalają się pod wpływem promieni słonecznych; spalanie się organicznych związków w chlorze charakteryzuje się wydzieleniem wolnego węgla w postaci sadzy: CH4 + 2Cl → 4HCl + C innym silnym utleniaczem jest nadmanganian potasu KmnO4, np. glikol etylowy w mieszaninie z nadmanganianem potasu ulega samozapaleniu już po 1-2 minutach.
, siarczki, węgliki alkalicznych metali, np: N2S). Wszystkie te substancje zdolne są utleniać się na powietrzu z wydzielaniem ciepła, kosztem którego reakcja przyspiesza się i następuje samonagrzewanie i w konsekwencji może nastąpić samozapalenie. Czas indukcji dla tych materiałów jest bardzo różny; • materiały, które ulegają samonagrzewaniu pod wpływem wilgoci; do tej grupy należą: sód, potas, rubid, cez, lit. ( pierwiastki I grupy układu okresowego), jak również tlenki metali alkalicznych. Wszystkie te substancje energicznie łączą się z wodą, wydziela się wówczas ciepło, które powoduje wzrost temperatury związku chemicznego i w konsekwencji samonagrzewanie, ewentualnie samozapalenie, np. w reakcji. 2Na + 2H2O → 2NaOH + H2. • materiały, które ulegają samonagrzewaniu w mieszaninach między sobą. Mieszaniny samozapalające otrzymuje się w wyniku zmieszania utleniacza z materiałem palnym. Najsilniejsze utleniacze to: kwas HNO3 i jego sole, szczególnie azotany I grupy układu okresowego. kwas siarkowy H2SO4. nadtlenki metali I i II grupy układu okresowego, np. N2O2. wszystkie tlenowe kwasy pochodne od chloru, np.KClO2. obok tlenu silnym utleniaczem jest chlor; acetylen, wodór, w mieszaninie z chlorem samozapalają się pod wpływem promieni słonecznych; spalanie się organicznych związków w chlorze charakteryzuje się wydzieleniem wolnego węgla w postaci sadzy: CH4 + 2Cl → 4HCl + C. innym silnym utleniaczem jest nadmanganian potasu KmnO4, np. glikol etylowy w mieszaninie z nadmanganianem potasu ulega samozapaleniu już po 1-2 minutach.")
186
Określenie zdolności do samonagrzewania się olejów roślinnych
Oleje dzieli się na: mineralne, roślinne i zwierzęce. Do samonagrzewania zdolne są tylko oleje roślinne. Oleje roślinne są to mieszaniny glicerydów wysoko cząsteczkowych kwasów tłuszczowych np.: kwasu stearynowego, palmitynowego czy też oleinowego. Glicerydy wyso kocząsteczkowych kwasów nasyconych (np: stearynowego) są substancjami stałymi i stanowią podstawowy składnik tłuszczów zwierzęcych. Glicerydy wysoko cząsteczkowych kwasów nienasyconych (np. oleinowego) są cieczami. To one stanowią podstawowy składnik olejów roślinnych. Cząsteczki tych glicerydów mają podwójne wiązania, które warunkują zdolność do samonagrzewania. Mechanizm utleniania olejów roślinnych Związki nienasycone, charakteryzują się dużą swobodną energią i dlatego w normalnych warunkach tlen cząsteczkowy zdolny jest do reakcji przyłączenia w miejscu podwójnych wiązań. Dzięki energii podwójnego wiązania jedno wiązanie w cząsteczce tlenu ulega rozerwaniu, w wyniku czego powstaje aktywna cząsteczka tlenu (― O ― O ―). Cząsteczka ta przyłącza się do grupy metylenowej (CH2), tworząc hydronadtlenek. R ― CH ≡ CH ― R + O → R ― CH ― CH ― R │ │ O ― O hydronadtlenek Nadtlenki i hydronadtlenki są związkami nietrwałymi. Przy ich rozkładzie tworzy się tlen atomowy, np: R1 ― CH2 ― CH ≡ C ― CH2 ―R2 → R1 ― CH2 ― CH = C ― C ― CH2 + O │ │ O ― O ― H O ― H
 są substancjami stałymi i stanowią podstawowy składnik tłuszczów zwierzęcych. Glicerydy wysoko cząsteczkowych kwasów nienasyconych (np. oleinowego) są cieczami. To one stanowią podstawowy składnik olejów roślinnych. Cząsteczki tych glicerydów mają podwójne wiązania, które warunkują zdolność do samonagrzewania. Mechanizm utleniania olejów roślinnych. Związki nienasycone, charakteryzują się dużą swobodną energią i dlatego w normalnych warunkach tlen cząsteczkowy zdolny jest do reakcji przyłączenia w miejscu podwójnych wiązań. Dzięki energii podwójnego wiązania jedno wiązanie w cząsteczce tlenu ulega rozerwaniu, w wyniku czego powstaje aktywna cząsteczka tlenu (― O ― O ―). Cząsteczka ta przyłącza się do grupy metylenowej (CH2), tworząc hydronadtlenek. R ― CH ≡ CH ― R + O → R ― CH ― CH ― R. │ │ O ― O. hydronadtlenek. Nadtlenki i hydronadtlenki są związkami nietrwałymi. Przy ich rozkładzie tworzy się tlen atomowy, np: R1 ― CH2 ― CH ≡ C ― CH2 ―R2 → R1 ― CH2 ― CH = C ― C ― CH2 + O. │ │ O ― O ― H O ― H.")
187
Powstały tlen atomowy jest bardzo reaktywny, dlatego też natychmiast wchodzi w reakcję utleniania, w wyniku czego podnosi się temperatura oleju. Wskaźnikiem oceny zdolności do samonagrzewania się olejów jest tzw. liczba jodowa ( jest to masa [g] jodu, którą może pochłonąć 100g oleju). Czym większa jest liczba jodowa oleju, tym więcej zawiera on związków nienasyconych i tym wyraźniejszą ma zdolność do utleniania się i samozapalenia. W poniższej tabeli podano liczby jodowe dla niektórych olejów: NAZWA OLEJU LICZBA JODOWA Lniany Słonecznikowy Konopiany Bawełniany Sojowy Rycynowy 82-86 Masło 25-47 Rybi Z powszechnie używanych olejów roślinnych olej lniany ma największą liczbę jodową, a więc największą zdolność do samozapalenia. Jeśli liczba jodowa oleju jest mniejsza od 89 samozapalenie nie nastąpi.
. Czym większa jest liczba jodowa oleju, tym więcej zawiera on związków nienasyconych i tym wyraźniejszą ma zdolność do utleniania się i samozapalenia. W poniższej tabeli podano liczby jodowe dla niektórych olejów: NAZWA OLEJU. LICZBA JODOWA. Lniany Słonecznikowy Konopiany Bawełniany Sojowy Rycynowy Masło Rybi Z powszechnie używanych olejów roślinnych olej lniany ma największą liczbę jodową, a więc największą zdolność do samozapalenia. Jeśli liczba jodowa oleju jest mniejsza od 89 samozapalenie nie nastąpi.")
188
Samonagrzewanie pod wpływem zjawisk fizycznych
Samonagrzewanie pod wpływem zjawisk fizycznych następuje przede wszystkim w wyniku adsorpcji gazów przez materiał palny, a następnie reakcji chemicznej (utleniania). Adsorpcja gazów daje efekty cieplne tylko wtedy, gdy materiałem palnym są zwykle węgle kamienne, tzn. takie, które mają w swym składzie ok. 60% węgla pierwiastkowego, a resztę stanowią domieszki typu rudy żelaza (nie utleniające się w normalnych warunkach), siarczki żelaza, siarczany żelazowe. Najbardziej podatnym składnikiem węgla na adsorpcję i utlenianie jest piryt, który ma wyjątkową zdolność do utleniania i samonagrzewania w niskich temperaturach. Szybkość utleniania węgla, a następnie szybkość samozapalenia, zależy m.in. od: a) początkowej temperatury, b) stężenia tlenu w otoczeniu, c) stopnia rozdrobnienia węgla.
. Adsorpcja gazów daje efekty cieplne tylko wtedy, gdy materiałem palnym są zwykle węgle kamienne, tzn. takie, które mają w swym składzie ok. 60% węgla pierwiastkowego, a resztę stanowią domieszki typu rudy żelaza (nie utleniające się w normalnych warunkach), siarczki żelaza, siarczany żelazowe. Najbardziej podatnym składnikiem węgla na adsorpcję i utlenianie jest piryt, który ma wyjątkową zdolność do utleniania i samonagrzewania w niskich temperaturach. Szybkość utleniania węgla, a następnie szybkość samozapalenia, zależy m.in. od: a) początkowej temperatury, b) stężenia tlenu w otoczeniu, c) stopnia rozdrobnienia węgla.")
189
Samonagrzewanie pod wpływem procesów biologicznych
Procesy biologiczne dotyczą produktów roślinnych, np. siana, koniczyny. Rośliny mogą ulec samonagrzewaniu w określonych warunkach, a mianowicie: gdy zostaną nagromadzone większe ilości materiału i gdy wegetacja komórek jeszcze się w nich nie zakończyła i pod wpływem bakterii zaczynają się procesy fermentacyjne i gnilne, dając początek miejscowemu samonagrzewaniu. Proces samonagrzewania produktów roślinnych można podzielić na 4 etapy. • Etap l. Pod wpływem wilgoci zaczynają się procesy wegetacyjne (oddychanie i rozmnażanie), w związku z tym wydziela się ciepło, gdyż procesy wegetacyjne są procesami egzotermicznymi. Temperatura produktu roślinnego podnosi się do 70°C. • Etap 2. Pod wpływem temperatury (70°C) giną mikroorganizmy, nie są więc dalej źródłem ciepła, w wyniku którego może nastąpić samonagrzewanie, jednakże niektóre substancje roślinne (białkowe pektyny) rozpadają się w temperaturze 70°C, tworząc tzw. żółty węgiel porowaty, charakteryzujący się silnymi własnościami adsorpcyjnymi. • Etap 3. W wyniku adsorpcji utleniacza przez węgiel temperatura wzrasta do ok. 130°C. Zaczynają się rozkładać różne elementy roślin, a w temperaturze 200°C rozkładają się wszystkie komórki roślinne. Tworzący się węgiel w wyniku rozpadu komórek roślinnych ze względu na duże „rozwinięcie" powierzchni ulega intensywnemu utlenianiu co powoduje wzrost temperatury do 280°C - 300°C. • Etap 4. Roślina ulega samozapaleniu.
, w związku z tym wydziela się ciepło, gdyż procesy wegetacyjne są procesami egzotermicznymi. Temperatura produktu roślinnego podnosi się do 70°C. • Etap 2. Pod wpływem temperatury (70°C) giną mikroorganizmy, nie są więc dalej źródłem ciepła, w wyniku którego może nastąpić samonagrzewanie, jednakże niektóre substancje roślinne (białkowe pektyny) rozpadają się w temperaturze 70°C, tworząc tzw. żółty węgiel porowaty, charakteryzujący się silnymi własnościami adsorpcyjnymi. • Etap 3. W wyniku adsorpcji utleniacza przez węgiel temperatura wzrasta do ok. 130°C. Zaczynają się rozkładać różne elementy roślin, a w temperaturze 200°C rozkładają się wszystkie komórki roślinne. Tworzący się węgiel w wyniku rozpadu komórek roślinnych ze względu na duże „rozwinięcie powierzchni ulega intensywnemu utlenianiu co powoduje wzrost temperatury do 280°C - 300°C. • Etap 4. Roślina ulega samozapaleniu.")
190
BLEVE – tragedia w San Juanico (MEXICO CITY)
► ranek r. - zakład dystrybucji gazu LPG, ► gęsto zaludniony obszar w odległości 300m od zakładu, ► pękł rurociąg o średnicy 200 mm, ► operator nie mógł porozumieć się z terminalem, skąd można było odciąć wypływ gazu, ► po 10 min olbrzymia chmura par LPG zapaliła się, ► po następnych 5 min pożar spowodował wybuch BLEVE dwóch dużych zbiorników kulistych (każdy zawierał 1500 m3 skroplonego gazu), ► kule ogniste osiągały 600 m, ► fragmenty zbiorników i instalacji o wadze ton lądowały w odległości 100 do 890 m od miejsca wybuchu, ► 15 z 48 cylindrycznych zbiorników (20 ton każdy), zmieniło się w pociski wznoszące się na wysokość 100m, po czym lądowały w odległości m ► temperatura : - w pobliżu zbiorników kulistych osiągnęła oC, - w płomieniu – fireball 1200 oC, - 200 m dalej – oC. ► na skutek pożaru i wybuchów spłonęło około m3 skroplonego gazu, ► zginęło 500 osób, 2000 odniosło poważne obrażenia (większość poparzenia), ► ludzi ewakuowano.
, ► kule ogniste osiągały 600 m, ► fragmenty zbiorników i instalacji o wadze ton lądowały w odległości 100 do 890 m. od miejsca wybuchu, ► 15 z 48 cylindrycznych zbiorników (20 ton każdy), zmieniło się w pociski wznoszące się na wysokość 100m, po czym lądowały w odległości m. ► temperatura : - w pobliżu zbiorników kulistych osiągnęła oC, - w płomieniu – fireball 1200 oC, m dalej – oC. ► na skutek pożaru i wybuchów spłonęło około m3 skroplonego gazu, ► zginęło 500 osób, 2000 odniosło poważne obrażenia (większość poparzenia), ► ludzi ewakuowano.")

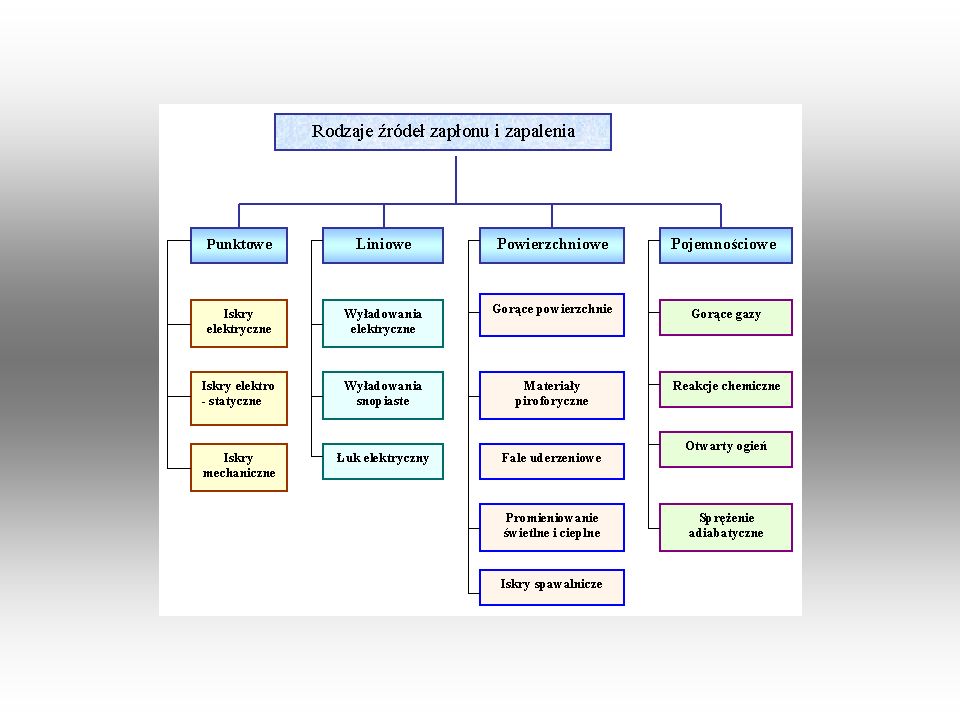
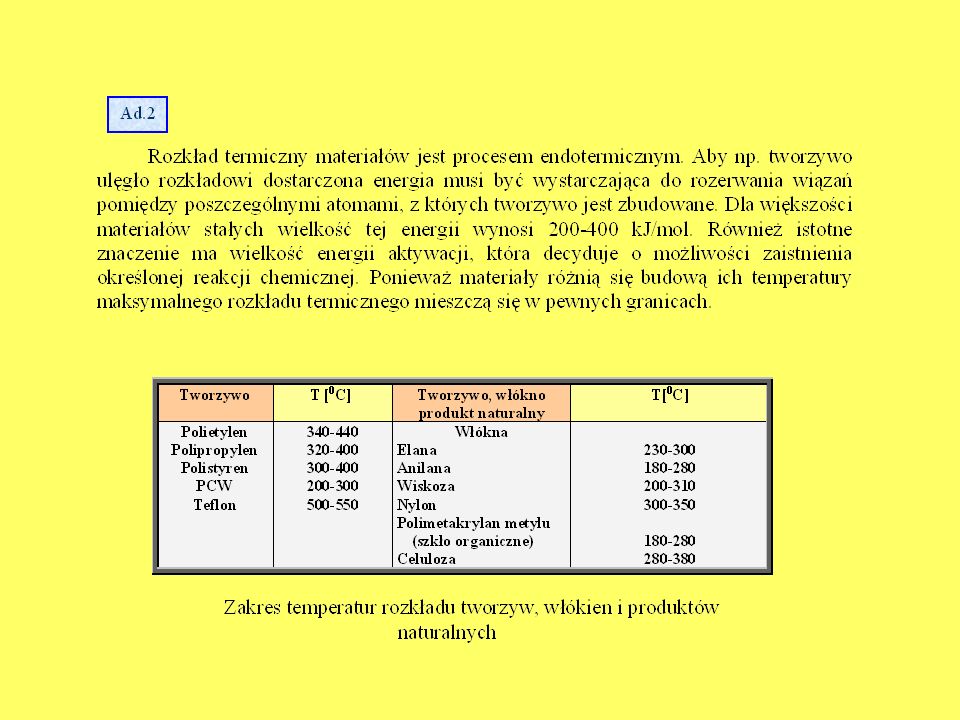
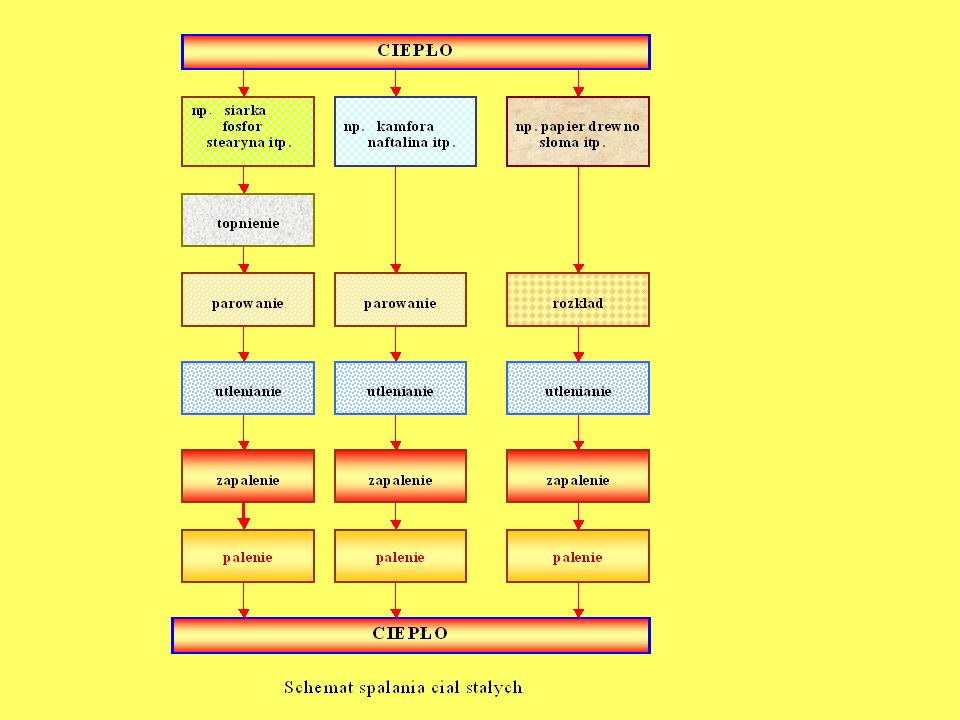
 ogół rzeczywistych jednostek, o których chcemy uzyskać informacje.>")
 rodzaj pracy,>")

 1. Energia mechaniczna 2. Praca 3.>")


1806 r. - J. Berzelius wprowadził nazwę „związki organiczne” dla wszystkich substancji występujących w organizmach roślinnych.>")
 1. Zjawisko tarcia 2. Tarcie ślizgowe.>")
 do zakresu komórek w innym skoroszycie Możliwości efektywnego stosowania odwołań zewnętrznych Odwołania zewnętrzne.>")





 (1623-1662) Blaise Pascal Ciśnienie wywierane na ciecz rozchodzi się jednakowo we wszystkich.>")


 Cechowanie (PN-EN )>")

