Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
WRODZONE WADY METABOLIZMU
„…what we learn from rare disorders often has profound consequences for our understanding of more common conditions.” Francis S. Collins Director of the National Human Genome Research Institute

2
DEFINICJA zaburzenia powstałe w wyniku genetycznie uwarunkowanych bloków enzymatycznych (mutacje w pojedynczym genie), prowadzące do nieprawidłowych procesów przemiany materii ponad 3000 wrodzonych wad metabolizmu
, prowadzące do nieprawidłowych procesów przemiany materii. ponad 3000 wrodzonych wad metabolizmu.")
3
CZĘSTOŚĆ WYSTĘPOWANIA
fenyloketonuria: 1:8000; galaktozemia: 1:40,000. choroba syropu klonowego 1:250,000 homocystynuria 1:250,000 inne 1:1,000,000

4
Najczęściej AR, pokrewieństwo rodziców ?
Wrodzone wady metabolizmu Inborn Errors of Metabolism (IEM) W Polsce rodzi się nowych przypadków rocznie, z czego ok jest rozpoznawanych Najczęściej AR, pokrewieństwo rodziców ? Pierwsze objawy kliniczne u noworodka w 2/3 przypadków IEM
 W Polsce rodzi się nowych przypadków rocznie, z czego ok jest rozpoznawanych. Najczęściej AR, pokrewieństwo rodziców Pierwsze objawy kliniczne u noworodka w 2/3 przypadków IEM.")
5
dziedziczenie mitochondrialne
SPOSÓB DZIEDZICZENIA AR AD sprzężone z X dziedziczenie mitochondrialne

6
TYPY WRODZONYCH WAD METABOLIZMU - I
Metabolizm aminokwasów fenyloketonuria, alkaptonuria, homocystynuria, choroba syropu klonowego Defekty cyklu mocznikowego Metabolizm wodorowęglanów metabolizm monosacharydów choroby spichrzeniowe glikogenu Metabolizm steroidów wrodzony przerost nadnerczy, brak wrażliwości na androgeny Metabolizm lipidów rodzinna hipercholesterolemia, Mukopolisacharoidozy zespół Hurlera, zespół Huntera, zespół Sanfilipo, zespół Morquio Sfingolipidozy choroba Tay-Sachsa, choroba Gaucher, choroba Niemann-Picka choroba Wilsona, choroba Menkesa

7
TYPY WRODZONYCH WAD METABOLIZMU - II
Metabolizm puryn i pirymidyn choroba Lesch-Nyhana Metabolizm porfiryn porfirie wątrobowe, erytropoetyczne Zaburzenia dot kwasów organicznych kwasica metylomalonowa, kwasica propionowa Metabolizm miedzi choroba Wilsona, choroba Menkesa Choroby peroksyzomalne zespół Zellwegera, adrenoleukodystrofia Choroby mitochondrialne Zaburzenia oksydacji kwasów tłuszczowych acyduria glutarowa

8
U chorego noworodka zawsze należy podejrzewać wadę metabolizmu
OBJAWY KLINICZNE U chorego noworodka zawsze należy podejrzewać wadę metabolizmu

9
Tabele na podstawie: Archives of disease in childhood fetal and neonatal edition. Chakrapani et al.., 2001;84;F205-F210

10
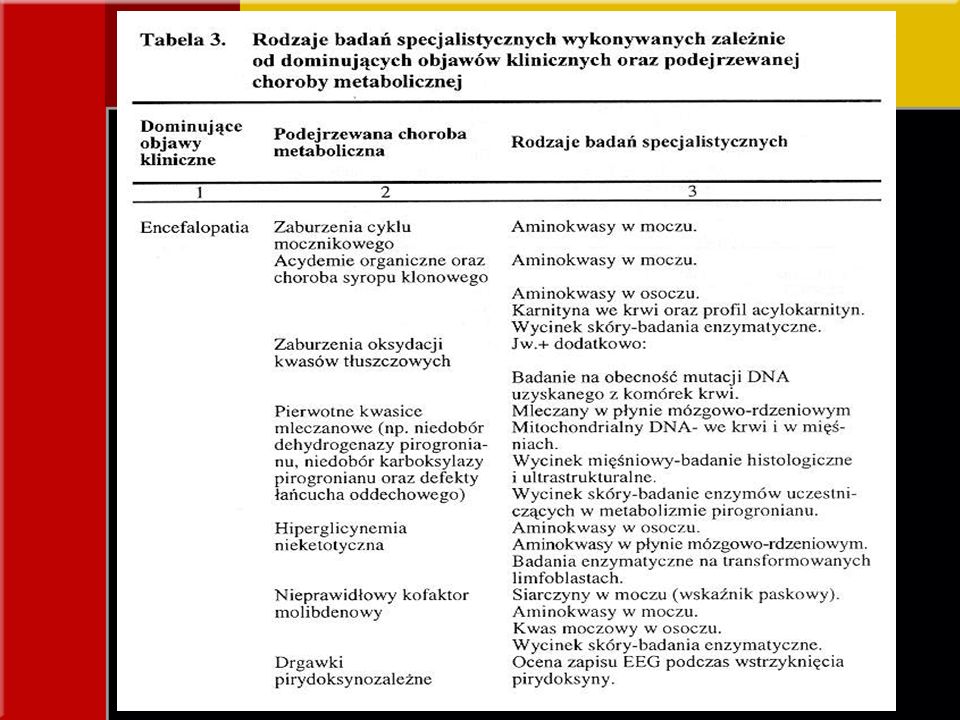
OBJAWY KLINICZNE Zaburzenia neurologiczne - encefalopatia i drgawki
*po urodzeniu zdrowe, w krótkim czasie senność, gorsze łaknienie, wymioty, niepokój, często wysokie stężenie NH3, zasadowica oddechowa, ketonuria acydurie organiczne: propionowa, metylomalonowa, izowalerianowa; zaburzenia cyklu mocznikowego *po urodzeniu utrata przytomności, drgawki, bezdech nieketotyczna hiperglicynemia, drgawki pirydoksynowe, niedobór kofaktora molibdenowego, pierwotna kwasica mleczanowa; choroby mitochondrialne i peroksyzomalne

12
OBJAWY KLINICZNE Kwasica metaboliczna Kwasica mleczanowa
przedłużająca się kwasica o niewyjaśnionej przyczynie zwiększona wartość luki anionowej acydemie organiczne Kwasica mleczanowa zaburzenia metabolizmu pirogronianów, defekty łańcucha oddechowego zaburzenia oksydacji kwasów tłuszczowych, kwasice organiczne, zaburzenia cyklu mocznikowego Stężenie mleczanów powyżej 3 mmol/l bez zamartwicy, uszkodzeń narządów - diagnostyka w kierunku wrodzonych wad metabolizmu

13
OBJAWY KLINICZNE Hipoglikemia
pobrać materiał do badań w trakcie epizodu hipoglikemii zaburzenia oksydacji tłuszczów, wąrobowe postacie glikogenoz, zaburzenia glukoneogenezy Wtórna hipoglikemia w chorobach metabolicznych pierwotnie zaburzających funkcje wątroby *po urodzeniu utrata przytomności, drgawki, bezdech nieketotyczna hiperglicynemia, drgawki pirydoksynowe, niedobór kofaktora molibdenowego, pierwotna kwasica mleczanowa; choroby mitochondrialne i peroksyzomalne

15
OBJAWY KLINICZNE Zaburzenia czynności wątroby
najczęstsza przyczyna metaboliczna - galaktozemia tyrozynemia wątrobowo-nerkowa niedobór alpha 1 – antytrypsyny hemochromatoza noworodków mitochondrialne zaburzenia łańcucha oddechowego typ C choroby Niemanna i Picka

17
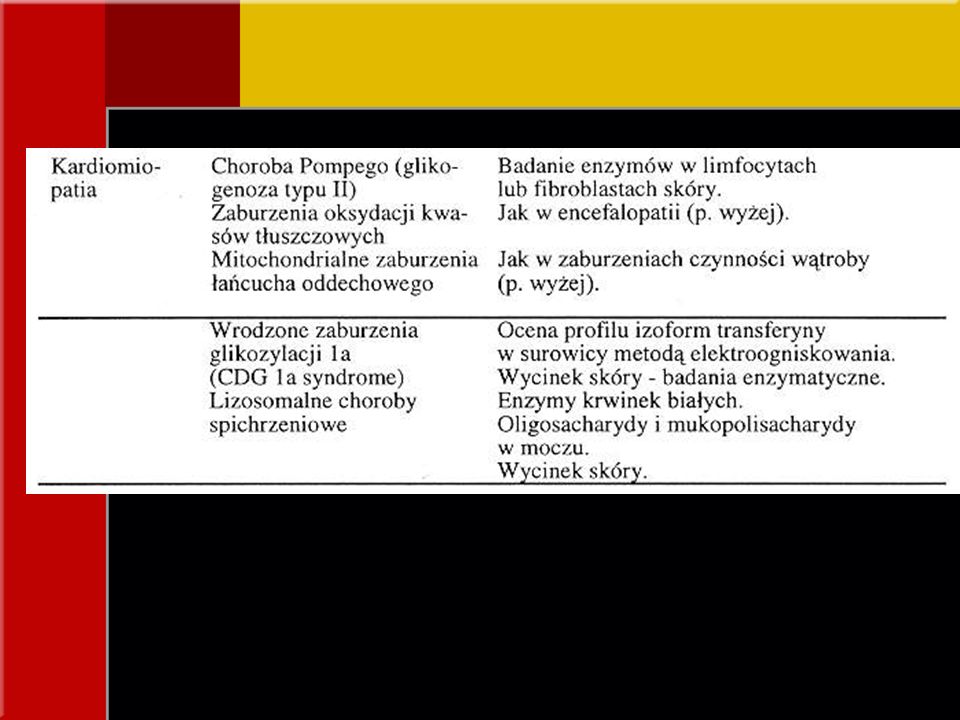
OBJAWY KLINICZNE Objawy ze strony serca
*niewydolność serca, kardiomiopatia przerostowa, hipotonia mitochondrialne zaburzenia łańcucha oddechowego zaburzenia oksydacji długołańcuchowych kwasów tłuszczowych, choroba Pompego (glikogenoza typ II) *kardiomiopatia i/lub płyn w osierdziu + zaburzenia rozwoju fizycznego, dysmorfia twarzy, wciągnięcie brodawek sutkowych, nieprawidłowe rozmieszczenie tkanki tłuszczowej wieloukładowe wrodzone zaburzenia glikozylacji (CDG) *kardiomiopatia rozstrzeniowa, neutropenia, zaburzenie stężenia kwasów organicznych w moczu (zespół Bartha)
 *kardiomiopatia i/lub płyn w osierdziu + zaburzenia rozwoju fizycznego, dysmorfia twarzy, wciągnięcie brodawek sutkowych, nieprawidłowe rozmieszczenie tkanki tłuszczowej. wieloukładowe wrodzone zaburzenia glikozylacji (CDG) *kardiomiopatia rozstrzeniowa, neutropenia, zaburzenie stężenia kwasów organicznych w moczu (zespół Bartha)")
19
OBJAWY KLINICZNE Dysmorfia
lizosomalne choroby spichrzeniowe - pogrubinie rysów twarzy zaburzenia syntezy steroli (SLOS) wrodzone zaburzenia glikozylacji acyduria glutarowa typu II, acyduria 3- hydroksyizomasłowa, tj. wpływające bezpośrednio na metabolizm energetyczny (wady serca, nerek, układu kostno-szkieletowego)
 wrodzone zaburzenia glikozylacji. acyduria glutarowa typu II, acyduria 3- hydroksyizomasłowa, tj. wpływające bezpośrednio na metabolizm energetyczny (wady serca, nerek, układu kostno-szkieletowego)")
20
OBJAWY KLINICZNE Inne choroba syropu klonowego
zapach spoconych stóp - kwasica izowalerianowa, acyduria glutarowa typu II niestabilność temperatury ciała – wczesny objaw zespółu Menkesa uogólniony nieimmunologiczny obrzęk płodu – wiele chorób metabolicznych żółtaczka, skaza krwotoczna – uszkodzenia wątroby, zaburzenia cyklu mocznikowego

23
BADANIA PRZESIEWOWE

24
Metabolizm aminokwasów

25
Fenyloketonuria mutacja w genie kodującym hydroksylazę fenyloalaniny PAH
toxic

26
Pierwsza wrodzona wada metabolizmu
Fenyloketonuria Pierwsza wrodzona wada metabolizmu rutynowo diagnozowana – podwyższone stężenie fenyloalaniny – podwyższone stężenie kw. fenylopirogronowego w moczu Opóźnienie rozwoju psychoruchowego - pierwszy objaw sugerujący chorobę. Z wiekiem dziecka opóźnienie rozwoju umysłowego zwykle w stopniu głębokim (iloraz inteligencji 20-40).
.")
27
OBJAWY FENYLOKETONURII
* wzrost fenyloalaniny (Phe) we krwi => uszkodzenie OUN * zmiany skórne (o charakterze zmian alergicznych lub zapalnych) * skłonność do wymiotów (sugerująca zwężenie odźwiernika) * wzmożona pobudliwość, spadek napięcia mięśniowego * wzmożenie odruchów ścięgnistych * drgawki (głównie pod postacią napadów zgieciowych) * zbyt wolne przyrosty obwodu głowy - małogłowe (60-90% chorych)! * opóźnienie rozwoju psychoruchowego * „rozcieńczenie barwnika" = jasna karnacja * "mysi zapach" => obecność kwasu ortohydroksyfenylooctowego w moczu pacjenta.
 we krwi => uszkodzenie OUN * zmiany skórne (o charakterze zmian alergicznych lub zapalnych) * skłonność do wymiotów (sugerująca zwężenie odźwiernika) * wzmożona pobudliwość, spadek napięcia mięśniowego * wzmożenie odruchów ścięgnistych * drgawki (głównie pod postacią napadów zgieciowych) * zbyt wolne przyrosty obwodu głowy - małogłowe (60-90% chorych)! * opóźnienie rozwoju psychoruchowego * „rozcieńczenie barwnika = jasna karnacja * mysi zapach => obecność kwasu ortohydroksyfenylooctowego w moczu pacjenta.")
28
Fenyloketonuria Warunkiem pozytywnych efektów - wprowadzenie diety w okresie noworodkowym. Podstawą diety - białkozastępcze preparaty nisko- lub bezfenyloalaninowe produkowane na bazie hydrolizatów białkowych lub pozbawione fenyloalaniny syntetyczne mieszaniny aminokwasów uzupełniane w witaminy, składniki mineralne, pierwiastki śladowe.

29
Fenyloketonuria Ważne kontynuowanie leczenia (diety niskofenyloalaninowej) u dziewcząt do wieku dorosłego i przez cały okres prokreacji. Dziecko urodzone przez matkę chorą na PKU która nie zachowuje niskiego poziomu fenyloalaniny jest narażone na : wady serca, mikrocefalię, upośledzenie umysłowe.

30
Metabolizm steroidów

31
Zespół Smitha-Lemlego-Opitza (SLOS)
AR mutacje w genie reduktazy 7-dehydrocholesterolu (DHCR7) - Cholesterol - synteza błon komórkowych, hormonów steroidowych istoty białej mózgu Częstość: 1:10,000 to 30,000; w Polsce, Słowacji, Czechach najwyższa częstość Objawy: liczne wady rozwojowe OUN (małogłowie), serca, zewnętrznych narządów płciowych, rozszczep podniebienia, zaćma, blepharoptoza, dodatkowe palce, syndaktylia Zahamowanie wzrostu, upośledzenie rozwoju psychofizycznego, zaburzenia zachowania
 - Cholesterol - synteza błon komórkowych, hormonów steroidowych istoty białej mózgu. Częstość: 1:10,000 to 30,000; w Polsce, Słowacji, Czechach. najwyższa częstość. Objawy: liczne wady rozwojowe OUN (małogłowie), serca, zewnętrznych narządów płciowych, rozszczep podniebienia, zaćma, blepharoptoza, dodatkowe palce, syndaktylia. Zahamowanie wzrostu, upośledzenie rozwoju psychofizycznego, zaburzenia zachowania.")
32
Zespół Smitha-Lemlego-Opitza (SLOS)
rozszczep podniebienia polidaktylia i bruzda poprzeczna obojnacze narządy płciowe syndaktylia drugiego i trzeciego palca

33
Upośledzona aktywność 7- redukatazy 7-dehydrocholesterolu, która katalizuje przemianę 7-dehydrocholesterolu (7-DHC) do cholesterolu i 7-dehydrocholesterolu do desmosterolu. Zahamowanie syntezy cholesterolu i zwiększone stężenie 7DHC w surowicy.

34
Zespół Smitha-Lemlego-Opitza (SLOS)
Badania biochemiczne krwi – podwyższone stężenie 7DHC. Poziom cholesterolu w surowicy krwi niski, ale u 10% mogą być wartości prawidłowe Analiza mutacji genu DHCR7, korelacja fenotyp-genotyp Badania w kierunku SLOS powinny być rozważone u płodów/dzieci z polidaktylią, syndaktylią, zaćmą, rozszczepem podniebienia, obojnaczymi narządami płciowymi i prawidłowym kariotypem.

35
Metabolizm monosacharydów

36
Defekt metabolizmu galaktozy częstość 1:60,000; AR
GALAKTOZEMIA Defekt metabolizmu galaktozy częstość 1:60,000; AR Mutacje w genach: galaktozo-1-fosfourydylotransferazy (GALT), UDP-galactozo-4-epimerazy (GALE) i galaktokinazy 1 (GALK1) Homozygota G/G GALT – aktywność enzymu poniżej 5% wartości prawidłowych Heterozygota G/N – aktywność enzymu około 50% wartości prawidłowych
, UDP-galactozo-4-epimerazy (GALE) i galaktokinazy 1 (GALK1) Homozygota G/G GALT – aktywność enzymu poniżej 5% wartości prawidłowych. Heterozygota G/N – aktywność enzymu około 50% wartości prawidłowych.")
37
GALAKTOZEMIA Objawy kliniczne mogą wystąpić w kilka godzin po pierwszym karmieniu: wymioty, brak przyrostu masy ciała, żółtaczka, skaza krwotoczna. powiększenie wątroby i śledziony, zaćma We krwi podwyższone stężenie galaktozy, obniżony poziom glukozy. W moczu: galaktozuria, białkomocz, aminoaciduria (obecność substancji redukujących)
")
38
GALAKTOZEMIA Leczenie: całkowite wykluczenie z diety galaktozy i laktozy.

39
Metabolizm sfingolipidów

40
Choroba Gauchera brak glukocerebrozydazy
enzym aktywny w lizosomach, rozkłada glukocerebrozydy do glukozy i ceramidów glukocerebrozydy obecne są w wielu tkankach, przede wszystkim w makrofagach szpiku, wątroby i śledziony glukocerebrozydy powstają w procesie rozkładu krwinek czerwonych, fagocytowanych i degradowanych przez makrofagi. Gromadzenie glukocerebrozydów w lizosomach upośledza funkcje makrofagów. Makrofagi + nierozłożone glukocerebrozydy - komórki Gauchera

41
Komórki Gauchera „łagodne” jądra komórkowe i
cytoplazma o wyglądzie pogniecionego papieru

42
Choroba Gauchera Typ I – dorosłych
Hepatosplenomegalia Cytopenia Choroba płuc Choroba kości (patologiczne złamania, bóle stawów i kości) Typ II – niemowlęca (nie obejmuje kości) między 3-6 mż słabe przyrosty masy ciała, hepatosplenomegalia, regresja rozwoju, wzmożone napięcie mięśniowe, drgawki, nawracające infekcje ukł. oddechowego, zgon w drugim roku życia
 Typ II – niemowlęca (nie obejmuje kości) między 3-6 mż słabe przyrosty masy ciała, hepatosplenomegalia, regresja rozwoju, wzmożone napięcie mięśniowe, drgawki, nawracające infekcje ukł. oddechowego, zgon w drugim roku życia.")
43
Choroba Gauchera - diagnostyka
Test fluorometryczny – ocena aktywności glukozylocrebrydazy w leukocyach krwi obwodowej wynik nie umożliwia przewidzenia ciężkości ani typu choroby Analiza mutacji potwierdza rozpoznanie kliniczne i wynik badań biochemicznych Badanie nosicielstwa w rodzinie probanda

44
Choroba Gauchera - postępowanie
Splenektomia Transfuzja krwi - niedokrwistość Leczenie p-bólowe Leczenie ortopedyczne Suplementacja bifosfonianów i wapnia – metabolizm kości. Enzymatyczna terapia zastępcza ( (Cerezyme ®), recombinowana glucozyloceramidaza zawierająca alpha-mannozyl , lepsza fagocytoza przez makrofagi. Regularne podania iv zmniejszyło nasilenie objawów hematologicznych, hepatosplenomegalię. Lek dobrze tolerowany Ok % pacientów produkuje p-ciała p-enzymowi Enzymatyczna terapia zastępcza choroba Fabryego, MPSI; Próby kliniczne choroba Pompego, MPS II, VI
, recombinowana glucozyloceramidaza zawierająca alpha-mannozyl , lepsza fagocytoza przez makrofagi. Regularne podania iv zmniejszyło nasilenie objawów hematologicznych, hepatosplenomegalię. Lek dobrze tolerowany. Ok % pacientów produkuje p-ciała p-enzymowi. Enzymatyczna terapia zastępcza choroba Fabryego, MPSI; Próby kliniczne choroba Pompego, MPS II, VI.")
45
WRODZONE WADY METABOLIZMU POSTĘPOWANIE
Ograniczenia dietetyczne metabolizm aminokwasów Witaminy np., suplementacja thiaminy w kwasicach melczanowych Dializa Enzymatyczna terapia zastępcza Terapia genowa Transplantacja narządów, szpiku Leczenie objawowe Diagnostyka prenatalna, preimplanatacyjna

46
Diseases Treated - Inborn Errors of Metabolism Adrenoleukodystrophy Bare-lymphocyte syndrome Dyskeratosis congenita Familial erythrophagocytic lymphohistiocytosis Gaucher disease Gunter disease Hunter syndrome Hurler syndrome (genetic) Inherited neuronal ceroid lipofuscinosis Krabbe disease Langerhans’-cell histiocytosis Lesch-Nyhan disease Leukocyte adhesion deficiency Osteopetrosis - genetic
 Inherited neuronal ceroid lipofuscinosis Krabbe disease Langerhans’-cell histiocytosis Lesch-Nyhan disease Leukocyte adhesion deficiency Osteopetrosis - genetic.")
Podobne prezentacje










 i WTÓRNE (NABYTE) NIEDOBORY ODPORNOŚCI>")




 wynikającą z defektu produkcji lub działania insuliny wydzielanej.>")






