Pobierz prezentację
1
Genetyka medyczna (WPROWADZENIE; KIERUNEK NEUROBIOLOGIA)
POLECANE PODRĘCZNIKI (do przygotowania do ćwiczeń i uzupełnienia treści wykładowych) Podstawy biologii komórki. B. Alberts, Wyd. Naukowe PWN, Wyd. 1. lub nowsze. Genetyka molekularna. Red.: Piotr Węgleński, PWN, Warszawa (Wydanie 2). lub nowsze Genomy, T.A. Brown, Wyd. Naukowe PWN, Warszawa 2001 Genetyka, Krótkie wykłady, P.C. Winter, G.I. Hickey, H.L. Fletcher, Wyd. Naukowe PWN, Warszawa 2000. Genetyka. Ilustrowany przewodnik. E. Passarge, Wyd. Lekarskie PZWL 2004 Biochemia, L. Stryer, Wyd. Naukowe PWN, Warszawa 2000 i nowsze. A.L. SIEROŃ
 Podstawy biologii komórki. B. Alberts, Wyd. Naukowe PWN, Wyd. 1. lub nowsze. Genetyka molekularna. Red.: Piotr Węgleński, PWN, Warszawa (Wydanie 2). lub nowsze. Genomy, T.A. Brown, Wyd. Naukowe PWN, Warszawa Genetyka, Krótkie wykłady, P.C. Winter, G.I. Hickey, H.L. Fletcher, Wyd. Naukowe PWN, Warszawa Genetyka. Ilustrowany przewodnik. E. Passarge, Wyd. Lekarskie PZWL Biochemia, L. Stryer, Wyd. Naukowe PWN, Warszawa 2000 i nowsze. A.L. SIEROŃ.")
2
Metody genetyki i biologii molekularnej WYKŁAD 1 (WPROWADZENIE DO GENETYKI KLNICZNEJ; KIERUNEK LEKARSKI, ROK III) PODZIAŁY KOMÓREK CYKL KOMÓRKOWY FAZY CYKLU REGULATORY CYKLU AKTYWATORY BLOKERY APOPTOZA SZLAKI MEJOZA A.L. SIEROŃ

3
Segregacja chromosomów
CYKL KOMÓRKOWY Segregacja chromosomów Podział komórki 2 Wielkość Komórki 1 2 (4C) Zawartość DNA 1 (2C) Replikacja DNA CDK1 activity Cyclin levels
 Zawartość. DNA. 1 (2C) Replikacja DNA. CDK1 activity. Cyclin levels.")
4
DNA JEDNONICIOWY MAJĄ GO NIEKTÓRE WIRUSY TYPU DNA CUKIER DEZOKSYRYBOZA
ZASADA AZOTOWA PIRYMIDYNA ZASADA AZOTOWA PURYNA RESZTA KWASU FOSFOROWEGO MAJĄ GO NIEKTÓRE WIRUSY TYPU DNA Aleksander L. Sieroń

5
DNA DWUNICIOWY MAJĄ GO WSZYSTKIE EUKARIOTA, BAKTERIE I WIĘKSZOŚĆ
CUKIER DEZOKSYRYBOZA MAJĄ GO WSZYSTKIE EUKARIOTA, BAKTERIE I WIĘKSZOŚĆ WIRUSÓW TYPU DNA ZASADA AZOTOWA PIRYMIDYNA ZASADA AZOTOWA PURYNA RESZTA KWASU FOSFOROWEGO C G A T Aleksander L. Sieroń

6
RNA JEDNONICIOWY MAJĄ GO WIRUSY TYPU RNA (RETROWIRUSY) CUKIER RYBOZA
ZASADA AZOTOWA PIRYMIDYNA ZASADA AZOTOWA PURYNA RESZTA KWASU FOSFOROWEGO MAJĄ GO WIRUSY TYPU RNA (RETROWIRUSY) URACYL ZA TYMINĘ Aleksander L. Sieroń
 URACYL. ZA. TYMINĘ. Aleksander L. Sieroń.")
7
DNA może „kształtować siebie” na różne sposoby aby „osiągnąć własne” cele w życiu. Struktura krystaliczna połączeń między dwiema jego formami dostarcza informacji o tym jak DNA może osiągać te „akrobatyczne” figury. Ryc. 1 | Nowy skręt. Struktura połączenia dwóch form B–Z opracowana przez Ha et al. (Ha, S. C., Lowenhaupt, K., Rich, A., Kim, Y. G. & Kim, K. K. Nature 437, 1183–1186 (2005).)1. Obszar lewoskrętny Z-DNA łączy się z prawoskrętną strukturą B-DNA poprzez złącze w którym jedna para zasad jest wykręcona na zewnątrz lub wystaje z heliksu DNA. (Ryc. zmodyfikowana z ref. 1.) Richard R. Sinden NATURE|Vol 437|20 October 2005
.)1. Obszar lewoskrętny Z-DNA łączy się z prawoskrętną strukturą B-DNA poprzez złącze w którym jedna para zasad jest wykręcona na zewnątrz lub wystaje z heliksu DNA. (Ryc. zmodyfikowana z ref. 1.) Richard R. Sinden NATURE|Vol 437|20 October")
8
Upakowanie DNA w nukleosomie
histony 2x [H2A, H2B, H3 i H4] (niebieskie i zielone) H1 (żółty) DNA 146 pz DNA między nukleosomami około 60 pz
 H1 (żółty) DNA. 146 pz. DNA między nukleosomami. około 60 pz.")
9
Schemat upakowania DNA w nukleosomie

10
Nić DNA Włókno Nukleosomalne Solenoid Supersolenoid Chromatyda Chromosom

11
Początek składanie/aktywacja
Początek składanie/aktywacja (białko) aktywna kohezja postępujące widełki replikacyjne czynniki Replikacji początek wyładowań hamowania
 aktywna. kohezja. postępujące. widełki replikacyjne. czynniki. Replikacji. początek. wyładowań. hamowania.")
12
Metody biologii molekularnej WYKŁAD 1 (WPROWADZENIE DO GENETYKI LEKARSKIEJ; KIERUNEK LEKARSKI, ROK III) CYKL KOMÓRKOWY FAZY CYKLU REGULATORY CYKLU AKTYWATORY BLOKERY APOPTOZA SZLAKI MEJOZA A.L. SIEROŃ

13
Cykl komórkowy 0,5 DO 24 GODZIN Bloker guzów geny, CDK Blokowanie
Czynniki wzrostu Oncogeny Cykliny & CDK Bloker guzów geny, CDK Blokowanie „Punkt zakazu” (Niemożliwość powrotu) Nowa komórka siostrzana Mitoza (podział komórek) Początek cyklu Cykl komórkowy 0,5 DO 24 GODZIN Synteza, (Podwojenie DNA)
 Nowa komórka siostrzana. Mitoza (podział komórek) Początek. cyklu. Cykl komórkowy. 0,5 DO 24 GODZIN. Synteza, (Podwojenie. DNA)")
14
Cykl komórkowy 0,5 DO 24 GODZIN Nowa komórka siostrzana
Mitoza (podział komórek) Regulacja przez mitochondria Początek cyklu Czynniki wzrostu Onkogeny Cykliny & CDK Cykl Krebsa (mitochondria) Bloker guzów geny, CDK Blokowanie Synteza, (Podwojenie DNA) „Punkt zakazu” (Niemożliwość powrotu) Cykl komórkowy 0,5 DO 24 GODZIN Aleksander L. Sieroń
 Regulacja przez mitochondria. Początek. cyklu. Czynniki wzrostu. Onkogeny. Cykliny & CDK. Cykl Krebsa. (mitochondria) Bloker guzów. geny, CDK. Blokowanie. Synteza, (Podwojenie. DNA) „Punkt zakazu (Niemożliwość powrotu) Cykl komórkowy. 0,5 DO 24 GODZIN. Aleksander L. Sieroń.")
15
MEJOZA CYKL KOMÓRKOWY APOPTOZA FAZY CYKLU SZLAKI REGULATORY CYKLU
AKTYWATORY BLOKERY APOPTOZA SZLAKI MEJOZA A.L. SIEROŃ

16
CYKL KOMÓRKOWY Segregacja chromosomów Podział komórki Replikacja DNA
2 Wielkość komórki 1 2 Zawartość DNA 1 Replikacja DNA Stężenie Cyklin Aktywność CDK (kinazy cyklino zależne – fosforylazy)
")
17
Cykl komórkowy jest regulowany przez CDK (kinazy zależne od cyklin).
POCZĄTEK CYKLU

18
Cykliny aktywują CDK POCZĄTEK CYKLU
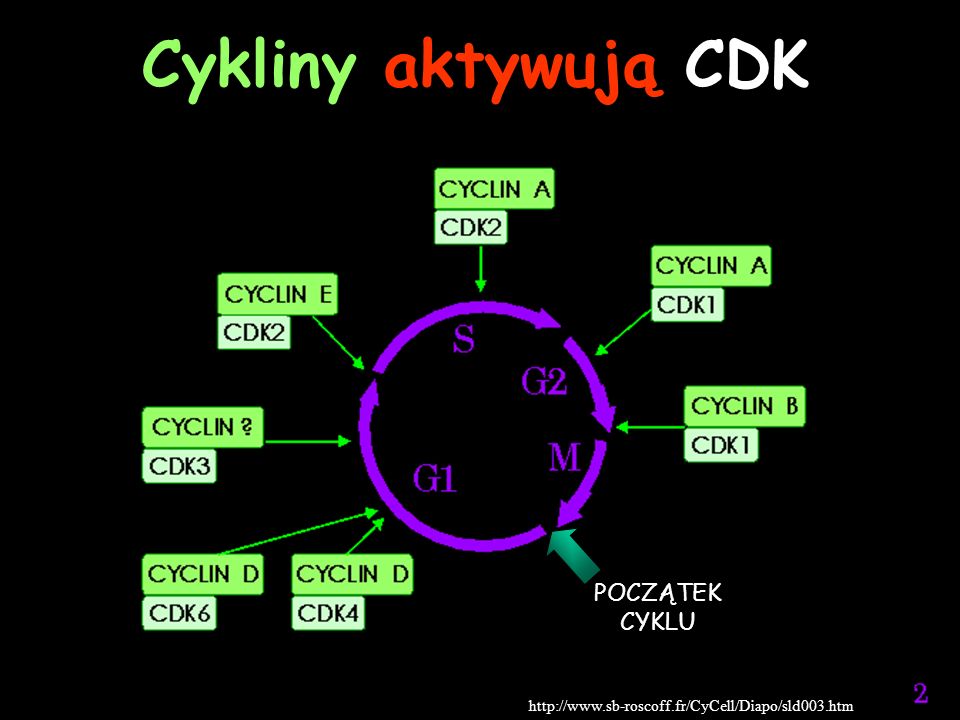
19
Strukturalny mechanizm
aktywacji Cdk Chapter 3 The Cell-Cycle Control System© 2007 New Science Press Ltd

20
Niskocząsteczkowe białka, p9cks i p15cdk-BP, i modulują ich aktywność
ściśle wiążą się z CDK i modulują ich aktywność POCZĄTEK CYKLU

21
Fosfatazy białkowe (cdc25) i inne kinazy (cdk7/cyklina H, polo)
Aktywatory CDK: Fosfatazy białkowe (cdc25) i inne kinazy (cdk7/cyklina H, polo) aktywują CDK. POCZĄTEK CYKLU
 i inne kinazy (cdk7/cyklina H, polo) aktywują CDK. POCZĄTEK CYKLU.")
22
MEJOZA CYKL KOMÓRKOWY APOPTOZA FAZY CYKLU SZLAKI REGULATORY CYKLU
AKTYWATORY BLOKERY APOPTOZA SZLAKI MEJOZA A.L. SIEROŃ

23
lub przez modyfikacje enzymatyczne (wee1, KAP, etc.)
Blokery CDK: Liczne białka blokują CDK wiążąc się z nimi stechiometrycznie (CIP1, INK4) lub przez modyfikacje enzymatyczne (wee1, KAP, etc.) POCZĄTEK CYKLU
 lub przez modyfikacje enzymatyczne (wee1, KAP, etc.) POCZĄTEK CYKLU.")
24
aktywny nieaktywny aktywny PROFAZA METAFAZA aktywny
Przejście profaza/metafaza: liczne enzymy utrzymują w profazie nieaktywny kompleks cdc2/cyklina B nieaktywny aktywny (G2) MITOZA (M) nieaktywny aktywny PROFAZA METAFAZA nieaktywny aktywny PROFAZA METAFAZA
 MITOZA (M) nieaktywny. aktywny. PROFAZA. METAFAZA. nieaktywny. aktywny. PROFAZA. METAFAZA.")
25
G2 MITOZA (M) nieaktywny aktywny Przejście profaza/metafaza:
Uszkodzenie DNA zatrzymuje komórki w profazie przez wpływ na aktywność cdc25 nieaktywny aktywny MITOZA (M) G2
 G2.")
26
Przejście profaza/metafaza: Inne czynniki regulatorowe!
MITOZA (M)
")
27
Oznaczanie aktywności kinazowej CDK1/cyklina B
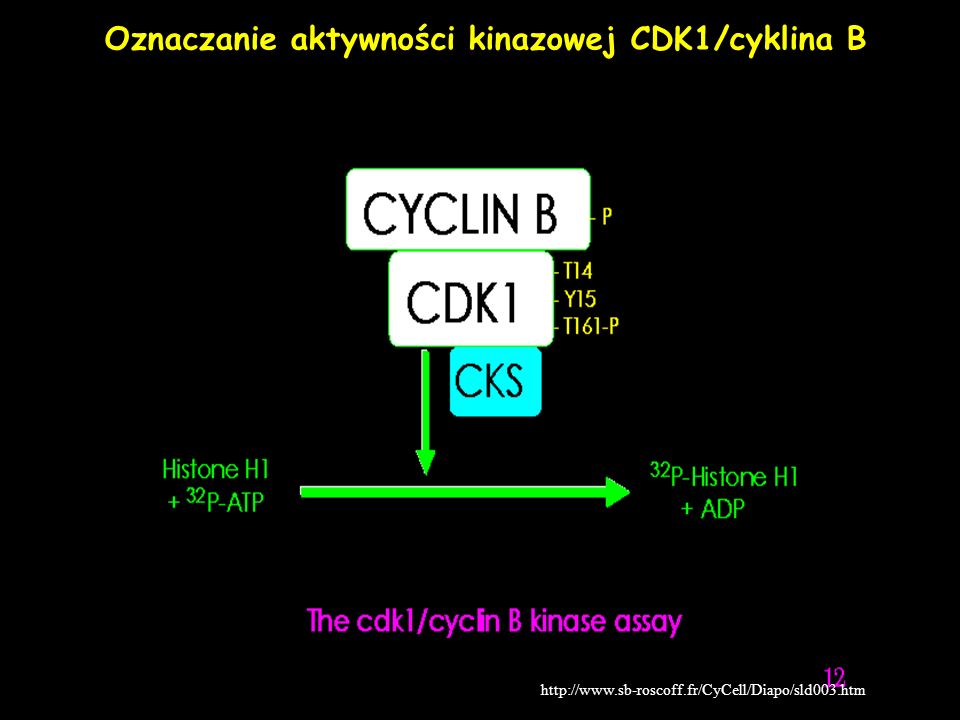
28
Potencjalne miejsca blokowania aktywności CDK
Miejsca fosforylowane Miejsca oddziaływania CDC25 Miejsca wiązania blokerów białkowych Kieszeń wiązania ATP Miejsca wiązania substratu Pętla fosfotreoniny Domena wiązania CKS CYKLIN „Skrzynka rozpadowa” Komórkowa lokalizacja domen

30
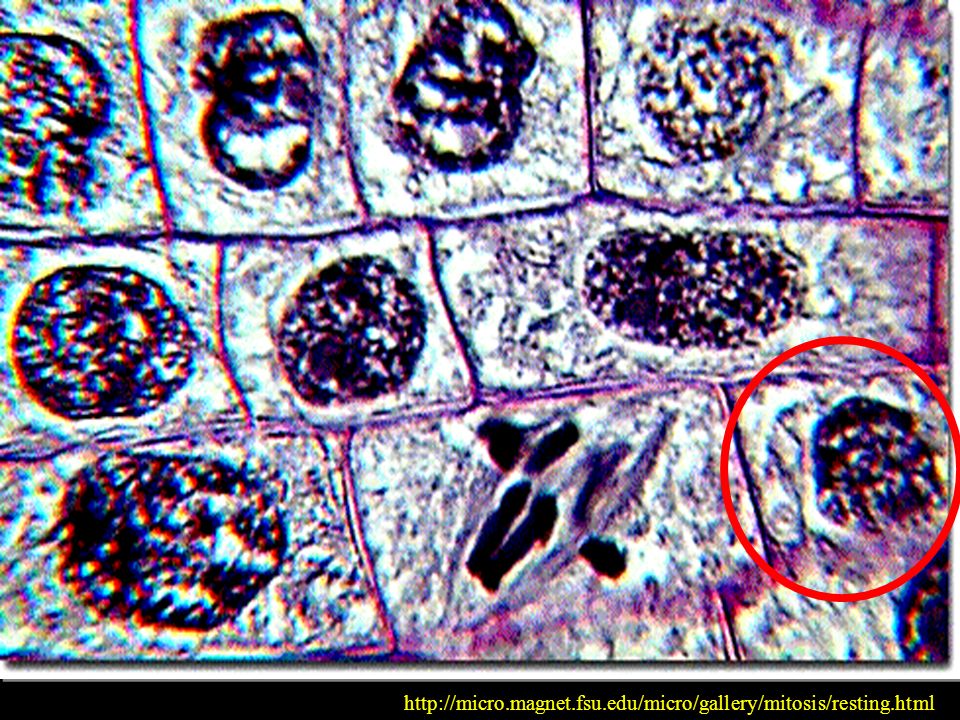
KOMÓRKA W SPOCZYNKU P O Z O R N I E !!!
32
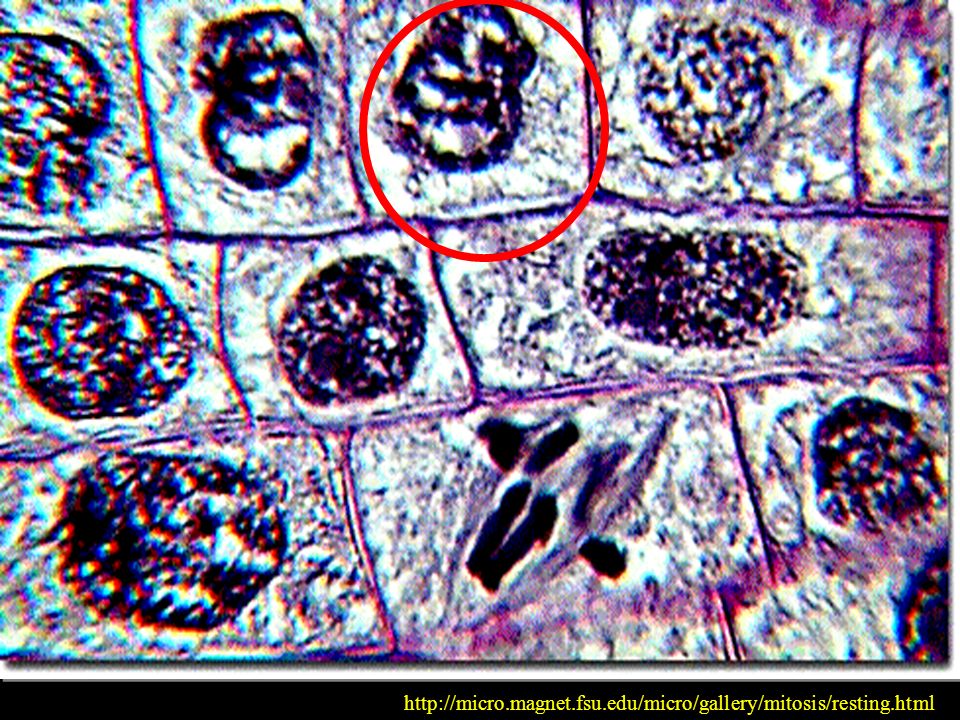
WCZESNA PROFAZA KONDENSACJA CHROMATYNY

34
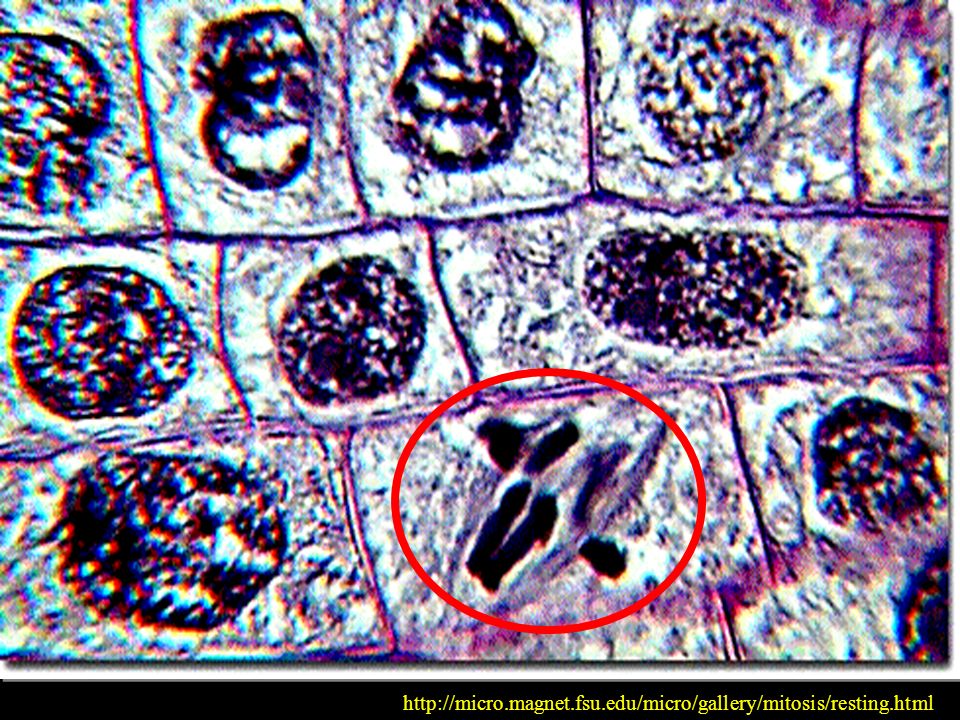
PÓŹNA PROFAZA CHROMOSOMY WĘDRUJĄ DO PŁYTKI METAFAZOWEJ

36
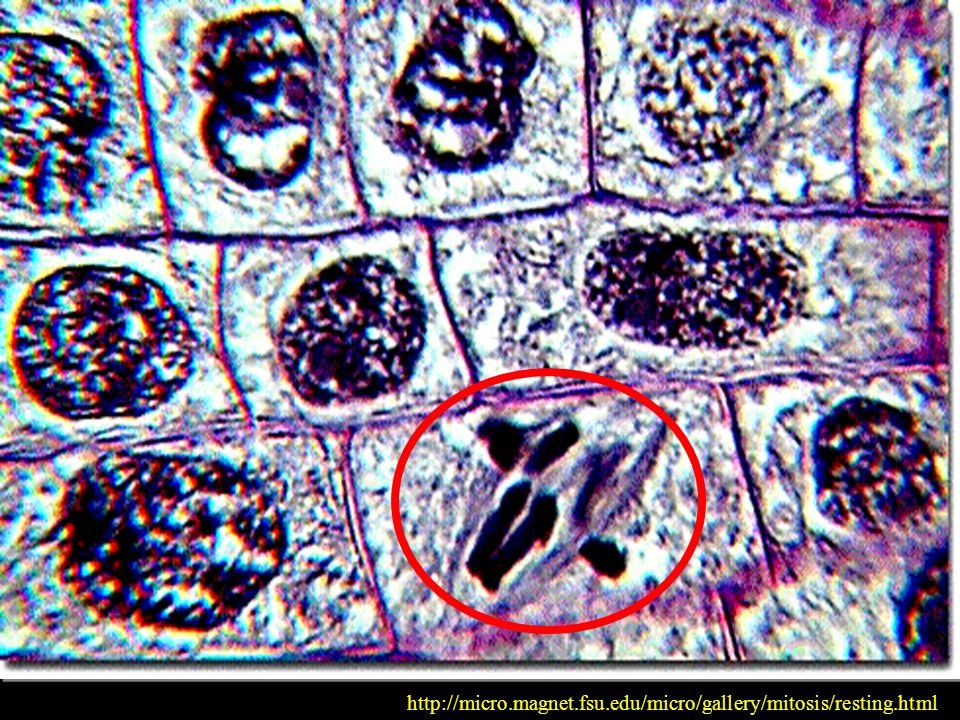
PÓŹNA METAFAZA CHROMOSOMY W PŁYTCE METAFAZOWEJ

38
Mitoza: Wczesna Anafaza
WĘDRÓWKA CHROMATYD DO BIEGUNÓW

39
MITOZA: PÓŹNA ANAFAZA DEKONDENSACJA CHROMATYNY

40
TELOFAZA CYTOKINEZA

41
KOMÓRKI POSTMITOTYCZNE
MITOZA: KOMÓRKI POSTMITOTYCZNE INTERFAZOWE JĄDRA

42
Partnerzy CDK

43
(natężenie cyklu Krebsa) (Różnicowanie komórek)
Na podstawie czynniki wzrostowe (natężenie cyklu Krebsa) i.t.p. faza S Cyklina D Cyklina E (Podziały komórek) (Różnicowanie komórek) (Naprawa komórek)
 i.t.p. faza S. Cyklina D. Cyklina E. (Podziały komórek) (Różnicowanie komórek) (Naprawa komórek)")
44
Wykrywanie uszkodzenia
Replikacja Uszkodzenie DNA Mocznik (HU) Wykrywanie uszkodzenia Faza S Odzysk „Zacisk” „Ładowacz zacisku”
 Wykrywanie uszkodzenia. Faza S. Odzysk. „Zacisk „Ładowacz zacisku")
45
Zatrzymanie cyklu komórkowego
Onkogeny (np. E1A, Myc) Uszkodzenie DNA (UV, leki) p19ARF Kinazy (np. ATM, DNAPK) p53 Zatrzymanie cyklu komórkowego lub apoptoza
 Uszkodzenie DNA. (UV, leki) p19ARF. Kinazy. (np. ATM, DNAPK) p53. Zatrzymanie cyklu komórkowego. lub apoptoza.")
46
Rb defosforylowane Cyklina B Cyklina A Cyklina D Cyklina E
Rb fosforylowane Na podstawie

47
(UV, PROMIENIOWANIE JONIZUJĄCE, LEKI, ITP.)
USZKODZENIE DNA (UV, PROMIENIOWANIE JONIZUJĄCE, LEKI, ITP.) p53 p21 *CYKLINA-CDK CYKLINA-CDK ATP ADP *E2F CYKLINA + CDK Rb:E2F ppRb G1 S
 p53. p21. *CYKLINA-CDK. CYKLINA-CDK. ATP. ADP. *E2F. CYKLINA + CDK. Rb:E2F. ppRb. G1. S.")
48
BRCA1 G2 M USZKODZENIA W KOMÓRCE ATM/ATR ? hCds1/Chk2
ATM, ATR i hCds1/Chk2 są białkami odpowiadającymi na uszkodznia w komórce zmieniając fosforylację produktu genu BRCA ATM/ATR ? hCds1/Chk2 BRCA1 Chk1 – Kinaza regulatorowa Cdc25C Kinaza Wee1 Cdc2/cyklina B G2 M

49
brak aktywności podziałowej
Linie kropkowane (krzyże) wskazują defekty w szlakach ATM i/lub ATR występujące w różnych liniach komórek rakowych. uszkodzenia DNA wczesna odpowiedź podtrzymanie brak aktywności podziałowej
 wskazują defekty w szlakach ATM i/lub ATR występujące w różnych liniach komórek rakowych. uszkodzenia DNA. wczesna odpowiedź. podtrzymanie. brak aktywności podziałowej.")
50
Cykl komórkowy cdk 1 cykliny A i B pRB/RIZ1 p53 p21 pRB cdk2, 4 i 6
Nowa komórka siostrzana Mitoza (podział komórek) Początek cyklu Czynniki wzrostu Oncogeny Cykliny & CDK Bloker guzów geny, CDK Blokowanie Synteza, (Podwojenie DNA) „Punkt zakazu” (Niemożliwość powrotu) Cykl komórkowy cdk 1 cykliny A i B pRB/RIZ1 Wejście do Apoptozy p53 p21 pRB cdk2, 4 i 6 cykliny A, E i D Wyjście do G0 Wejście do Apoptozy
 Początek. cyklu. Czynniki wzrostu. Oncogeny. Cykliny & CDK. Bloker guzów. geny, CDK. Blokowanie. Synteza, (Podwojenie. DNA) „Punkt zakazu (Niemożliwość powrotu) Cykl komórkowy. cdk 1. cykliny A i B. pRB/RIZ1. Wejście. do. Apoptozy. p53. p21. pRB. cdk2, 4 i 6. cykliny A, E i D. Wyjście. do G0. Wejście. do. Apoptozy.")
51
? Uszkodzenie DNA Zanik Telomerów Onkogeny
Model apoptotycznej odpowiedzi komórek na cytotoksyny, napromieniowanie i niedobór cytokin. Białka BH3-tylko Puma, Noxa i Bim pośredniczą, zarówno w zależnej, jak i niezależnej od p53 odpowiedzi na stres. Odpowiedzi te są aktywowane jednakowo przez stres wymuszony (niedobór cytokin, cytotoksyny i promieniowanie), jak i sygnały o stresie, takie które powstają w czasie tumorogenezy od aktywowanych onkogenów, zaniku telomerów i w hypoksji. Aktywacja kinazy ATM lub białka Arf blokuje białko Mdm2, co wywołuje wzrost stężenia p53 w komórkach. P53 indukuje transkrypcję genów p21, Puma i Noxa, a także innych o słabo poznanym znaczeniu (pola zakreskowane). Aktywowanie Bcl-2 (i innych pokrewnych mu białek) przez białka BH3-tylko powoduje aktywację Bax i Bak co wzbudza apoptozę. Skróty: STS, staurosporyna; dex, deksametazon. ATM Arf ? Mdm2 ? Hipoksja p53 Niedobór cytokin Wypływ Ca2+ p21 Taksol PMA Zatrzymanie Cyklu komórkowego STS dex PIG3, PIG8, PERP P53AIP1, p53DINP1 Bim Puma Noxa ? Bcl-2 Bax/ Bak Villunger A. et al. (2003) Science, 302: Apoptoza
, jak i sygnały o stresie, takie które powstają w czasie tumorogenezy od aktywowanych onkogenów, zaniku telomerów i w hypoksji. Aktywacja kinazy ATM lub białka Arf blokuje białko Mdm2, co wywołuje wzrost stężenia p53 w komórkach. P53 indukuje transkrypcję genów p21, Puma i Noxa, a także innych o słabo poznanym znaczeniu (pola zakreskowane). Aktywowanie Bcl-2 (i innych pokrewnych mu białek) przez białka BH3-tylko powoduje aktywację Bax i Bak co wzbudza apoptozę. Skróty: STS, staurosporyna; dex, deksametazon. ATM. Arf. Mdm2. Hipoksja. p53. Niedobór. cytokin. Wypływ Ca2+ p21. Taksol. PMA. Zatrzymanie. Cyklu komórkowego. STS. dex. PIG3, PIG8, PERP. P53AIP1, p53DINP1. Bim. Puma. Noxa. Bcl-2. Bax/ Bak. Villunger A. et al. (2003) Science, 302: Apoptoza.")
52
punkt kontrolny wymagane dla wyjścia z mitozy, które ma miejsce
Na podstawie punkt kontrolny meta anafaza cytokiny fazy S wymagane dla wyjścia z mitozy, które ma miejsce w fazach G1 i G2 cyklu komórkowego

53
kaskada kinazowa jąderko wyjście z mitozy
Na podstawie kaskada kinazowa jąderko wyjście z mitozy

54
kinaza naprawa faza S faza M i
uszkodzenie we wczesnej fazie S uszkodzenie w późnej fazie S/G2 kinaza naprawa faza S faza M i

55
Wejście do anafazy Wyjście z mitozy Zatrzymanie widełek Zatrzymanie
Wejście do anafazy Wyjście z mitozy Zatrzymanie widełek Zatrzymanie widełek

56
Uszkodzenia wrzeciona
Uszkodzenia wrzeciona kinaza meta metafaza

57
APC CDC20 HCT1/CDH1 meta anafaza
Na podstawie APC CDC20 HCT1/CDH1 meta anafaza cytokiny fazy S wyjście z mitozy

58
typ dziki uszkodzenie naprawa komórek nie może przywrócić
uszkodzenie typ dziki zmutowane komórki w punkcie kontrolnym nie mogą zatrzymać podziałów mikrokolonii naprawa komórek nie może przywrócić pojedynczych zmutowanych komórek

59
MEJOZA CYKL KOMÓRKOWY APOPTOZA FAZY CYKLU SZLAKI REGULATORY CYKLU
AKTYWATORY BLOKERY APOPTOZA SZLAKI MEJOZA A.L. SIEROŃ

60
ŚMIERĆ KOMÓRKI I APOPTOZA
Śmierć komórek następuje przez jeden z dwóch mechanizmów: 1. NEKROZĘ - następującą w wyniku działania uszkadzających czynników zewnętrznych np.: ciężkie oparzenia urazy mechaniczne 2. APOPTOZĘ - czyli zaprogramowaną śmierć komórek - będącą wynikiem następujących kolejno po sobie reakcji na bodźce biochemiczne lub fizyczne.

61
APOPTOZA Jest złożonym sposobem pozbywania się przez organizm niechcianych lub uszkodzonych komórek. Jest zaangażowana w homeostazę tkanek i różnicowanie komórek. Większość komórek organizmu ma z góry określoną długość życia. Śmierć komórki jest normalnie, ściśle regulowanym procesem, w którym komórki nieustannie odpowiadają na sygnały chemiczne pochodzące od innych komórek lub ze swojego otoczenia.

62
APOPTOZA jest bezpośrednio zaangażowana
w degeneracji; np. choroby: Alzheimera, Huntigtona, Parkinsonizm. w autoimmunoagresji: odczyny reumatyczne w choroby wirusowe: AIDS w przemianach nowotworowych

63
S T A R T Apoptoza zaczyna się w komórkach nie dających się naprawić, lub które zakończyły swoją zaprogramowaną funkcję biologiczną. Inna morfologia komórek wynika z zaburzeń w budowie błon Komórkowych.

64
WYKŁAD 1 cz. 2 (BIOLOGIA I BIOLOGIA Z GENETYKĄ; KIERUNKI: LEKARSKI, III ROK)
CYKL KOMÓRKOWY FAZY CYKLU REGULATORY CYKLU AKTYWATORY BLOKERY APOPTOZA GRACZE SZLAKI MEJOZA A.L. SIEROŃ

65
GRACZE W APOPTOZIE

66
DOMENA ZEWNĄTRZ KOMÓRKOWA
Complex DOMENA BŁONOWA DOMENA ZEWNĄTRZ KOMÓRKOWA CYTOPLAZMATYCZNA DOMENA

67
Complex ZEWNĄTRZ CYTOPLAZMA

68
WYKŁAD 1 cz. 2 (BIOLOGIA I BIOLOGIA Z GENETYKĄ; KIERUNKI: LEKARSKI, III ROK)
CYKL KOMÓRKOWY FAZY CYKLU REGULATORY CYKLU AKTYWATORY BLOKERY APOPTOZA GRACZE SZLAKI MEJOZA A.L. SIEROŃ

69
SZLAKI APOPTOZY RECEPTOROWY A P O P T O Z A MITOCHONDRIALNY

70
Szlak mitochondrialny
Szlak receptorowy FasL Domena zewnątrz-komórkowa Fas/CD95 Błona komórkowa Szlak mitochondrialny pro--Bid cytochrom c kaspaz-9/cytochrom c Kaspazy wykonawcze & inne substraty mitochondria p15tBid FADD pro-kaspaza-8

71
ODCZYNY ZAPALNE (NEKROZA)
(Cys-Proteazy) ODCZYNY ZAPALNE (NEKROZA) A P O P T O Z A
 ODCZYNY ZAPALNE (NEKROZA) A P O P T O Z A.")
72
A P O P T O Z A GRACZE W APOPTOZIE ODCZYNY ZAPALNE (NEKROZA)
Szlak mitochondrialny Szlak receptorowy ODCZYNY ZAPALNE (NEKROZA) A P O P T O Z A
 A P O P T O Z A.")
73
Po przyłączeniu ligandu (np..: Fas)
Szlak receptorowy Domena zewnątrz-komórkowa FasL Fas/CD95 Błona komórkowa Po przyłączeniu ligandu (np..: Fas) Szlak mitochondrialny FADD cytochrom c kaspaza-9/cytochrom c Kaspazy wykonawcze & inne substraty mitochondria p15tBid pro--Bid pro-kaspaza-8 pro-kaspaza-8 kaspaza 8
 Szlak mitochondrialny. FADD. cytochrom c. kaspaza-9/cytochrom c. Kaspazy wykonawcze & inne substraty. mitochondria. p15tBid. pro--Bid. pro-kaspaza-8. pro-kaspaza-8. kaspaza 8.")
74
Szlak mitochondrialny
Szlak receptorowy pro-kaspaza-8 pro--Bid kaspaza 8 cytochrom c kaspaza-9/cytochrom c mitochondria p15tBid Domena zewnątrz-komórkowa FasL Fas/CD95 Błona komórkowa FADD Szlak mitochondrialny kaspaza 3 Kaspazy wykonawcze & inne substraty

75
MUTACJE NIEAKTYWNE LIGANDY
pro-kaspaza-8 Domena zewnątrz-komórkowa FasL Fas/CD95 Błona komórkowa FADD Jądro komórkowe Bcl2 MUTACJE NIEAKTYWNA KASPAZA 8 MUTACJE NIEAKTYWNE RECEPTORY p53 Bax MUTACJE NIEAKTYWNE KINAZY Kaspazy wykonawcze & inne substraty kaspaza 3 cytochrom c kaspaz-9/cytochrom c p15tBid mitochondria

76
Rycina 6 Model wpływu Bcl-xS w fibroblastach zarodków mysich (MEFs).
Lindenboim L., et al. Cell Death and Differentiation (2005) 12, 713–723 Rycina 6 Model wpływu Bcl-xS w fibroblastach zarodków mysich (MEFs). Ekspresja Bcl-xS w MEFs wzbudza ekspozycję N-końca (NT) Bak, co prowadzi do aktywacji Bak. Zaktywowany Bak może indukować as trzy ścieżki sygnałowe: główna ścieżka prowadzi do uwolnienia cytochromu c, który aktywuje apoptosom i nastepczą śmierć komórki na drodze zależnej od kaspaz; druga ścieżka, prowadzi do Apaf-1- i niezależnej od kaspazy-9 śmierci komórkie; a trzecia ścieżka wzbudza ekspozycję NT Nax aktywując go na obydwu drogach zależnej- i niezależnej od caspazy-9. Pierwsze dwie ścieżki mają udział w procesach śmierci. Znaczenie trzeciej ścieżki jest dotychczas słabo poznane.
 12, 713–723. Rycina 6 Model wpływu Bcl-xS w fibroblastach zarodków mysich (MEFs). Ekspresja Bcl-xS w MEFs wzbudza ekspozycję N-końca (NT) Bak, co prowadzi do aktywacji Bak. Zaktywowany Bak może indukować as trzy ścieżki sygnałowe: główna ścieżka prowadzi do uwolnienia cytochromu c, który aktywuje apoptosom i nastepczą śmierć komórki na drodze zależnej od kaspaz; druga ścieżka, prowadzi do Apaf-1- i niezależnej od kaspazy-9 śmierci komórkie; a trzecia ścieżka wzbudza ekspozycję NT Nax aktywując go na obydwu drogach zależnej- i niezależnej od caspazy-9. Pierwsze dwie ścieżki mają udział w procesach śmierci. Znaczenie trzeciej ścieżki jest dotychczas słabo poznane.")
77
PRZEBIEG APOPTOZY W jądrze komórkowym („mózgu” komórki) chromatyna ulega zagęsz-czeniu, a DNA fragmentacji. Komórka w apoptozie jest otaczana przez sąsiadujące normalne komór-ki, które pochłaniają jej fragmenty i zużywają je na własne potrzeby.

78
Główne ścieżki prowadzące do śmierci kaspozo -zależnej i –niezależnej
Główne ścieżki prowadzące do śmierci kaspozo -zależnej i –niezależnej. Zidentyfikowano dwie ścieżki apoptotyczne kaspazo-zależne: ścieżkę zewnętrzną pobudzaną przez czynniki należące do nadrodziny typu receptora TNF-NGF (czynnik wzrostu nerwu), takie jak receptor TNF (TNFR), CD95 (Fas)-APO-1 receptor lub receptor typu TRAIL (‘receptory śmierci’) oraz ścieżkę wewnętrzną z udziałem MOMP prowadzącym do formowania kompleksów aktywujących kaspazy pomiędzy kaspazą 9 i Apaf-1 (apoptosom). Pobudzenie receptora śmierci prowadzi zwykle do rekrutacji i aktywacji kaspazy 8 za pośrednictwem białek adaptorowych FADD i TRADD, tworzących DISC, który rozprzestrzenia sygnał śmierci dwiema drogami: przez proteolizę białka BH3-only protein Bid, co wywołuje przemieszczenie tego ostatniego do mitochondrium i MOMP oraz przez bezpośrednią proteolizę następnych kaspaz, która powoduje ich aktywację. Na ścieżce wewnętrznej białka BH3-only działają tylko w odpowiedzi na stres komórkowy, uszkodzenie lub infekcję i mogą być mobilizowane do pobudzania MOMP w drodze modyfikacji potranslacyjnych. Białka BH3-only najprawdopodobniej wzbudzają MOMP poprzez zapoczątkowanie oligomeryzacji Bax i/lub Bak w zewnętrznej błonie mitochondrialnej, w której tworzą kanały, przez które uciekają liczne białka z przestrzeni międzybłonowej. W odniesieniu do uszkodzenia DNA stabilizacja białka supresora guzów p53 może prowadzić do aktywacji transkrypcji białek BH3-only, Puma i Noxa promujących MOMP poprzez kanał Bax-Bak. Alternatywna ścieżka apoptozy zależnej od p53 proponuje mechanizm z udziałem transkrypcyjnej nadregulacji białka PIDD. PIDD może promować tworzenie kompleksu własnego z RAIDD i kaspazą 2 (‘piddosomu’). Nie wiadomo dokładnie, jak piddosom może promować śmierć komórki, ale może w tym procesie uczestniczyć MOMP zależny od kaspazy 2. Niektóre białka mitochondrialne uwolnione przez MOMP (AIF, HtrA2/Omi, endonukleaza G) mogą promować śmierć komórki niezależną od kaspazy poprzez jeszcze bardzo słabo poznany mechanizm. Śmierć komórki niezależna od kaspaz może być także wynikiem pobudzeń prowadzących do zwiększonej przepuszczalności błony lizosomalnej (LMP) i zwiększonego uwalniania proteaz katepsynowych.
, takie jak receptor TNF (TNFR), CD95 (Fas)-APO-1 receptor lub receptor typu TRAIL (‘receptory śmierci’) oraz ścieżkę wewnętrzną z udziałem MOMP prowadzącym do formowania kompleksów aktywujących kaspazy pomiędzy kaspazą 9 i Apaf-1 (apoptosom). Pobudzenie receptora śmierci prowadzi zwykle do rekrutacji i aktywacji kaspazy 8 za pośrednictwem białek adaptorowych FADD i TRADD, tworzących DISC, który rozprzestrzenia sygnał śmierci dwiema drogami: przez proteolizę białka BH3-only protein Bid, co wywołuje przemieszczenie tego ostatniego do mitochondrium i MOMP oraz przez bezpośrednią proteolizę następnych kaspaz, która powoduje ich aktywację. Na ścieżce wewnętrznej białka BH3-only działają tylko w odpowiedzi na stres komórkowy, uszkodzenie lub infekcję i mogą być mobilizowane do pobudzania MOMP w drodze modyfikacji potranslacyjnych. Białka BH3-only najprawdopodobniej wzbudzają MOMP poprzez zapoczątkowanie oligomeryzacji Bax i/lub Bak w zewnętrznej błonie mitochondrialnej, w której tworzą kanały, przez które uciekają liczne białka z przestrzeni międzybłonowej. W odniesieniu do uszkodzenia DNA stabilizacja białka supresora guzów p53 może prowadzić do aktywacji transkrypcji białek BH3-only, Puma i Noxa promujących MOMP poprzez kanał Bax-Bak. Alternatywna ścieżka apoptozy zależnej od p53 proponuje mechanizm z udziałem transkrypcyjnej nadregulacji białka PIDD. PIDD może promować tworzenie kompleksu własnego z RAIDD i kaspazą 2 (‘piddosomu’). Nie wiadomo dokładnie, jak piddosom może promować śmierć komórki, ale może w tym procesie uczestniczyć MOMP zależny od kaspazy 2. Niektóre białka mitochondrialne uwolnione przez MOMP (AIF, HtrA2/Omi, endonukleaza G) mogą promować śmierć komórki niezależną od kaspazy poprzez jeszcze bardzo słabo poznany mechanizm. Śmierć komórki niezależna od kaspaz może być także wynikiem pobudzeń prowadzących do zwiększonej przepuszczalności błony lizosomalnej (LMP) i zwiększonego uwalniania proteaz katepsynowych.")
79
Shin S. et al. EMBO J. (2005) 24, 3532–3542 Proponowany mechanizm regulacji apoptozy z udziałem TRAIL. (Lewy schemat) Niska aktywność wewnątrzkomórkowego PKCK2 (1) lub wysoka aktywność wewnątrzkomórkowego PKCK2 jest obniżana przez specyficzny inhibitor (1’), defosforylowane monomery prokaspazy-2. Prokaspaza-2 jest następnie aktywowana w wyniku jej dimeryzacji (2), a zaktywowana kaspaza-2 tnie monomer prokaspazy-8 pomiędzy większą i mniejszą podjednostką (3). W takiej sytuacji dochodzi do ‘wzbudzenia’ apoptozy zależnej od TRAIL w komórkach nowotoworowych. Jeżeli TRAIL nie jest połączony z receptorami TRAIL-śmierć, cięta prokaspaza-8 jest kierowana do proteasomu celem degradacji (4). W przypadku przeciwnym TRAIL jest wiązany do swego receptora, przecięta prokaspaza-8 jest rekrutowana przez receptory śmierci TRAIL, czego wynikiem jest utworzenie DISC (4’). Drugie cięcie między prodomeną i podjednostką większą może być wykonane efektywnie w wyniku dimeryzacji ciętych prokaspaz-8, która zachodzi z udziałem DISC (5), co prowadzi do aktywacji prokaspazy-8 (6), a następnie apoptozy za pośrednictwem TRAIL (7). (Prawy schemat) Gdy wewnątrzkomórkowa aktywność PKCK2 jest wysoka prokaspaza-2 nie może być aktywowana; a zatem prokaspaza-8 nie może być przekształcana. Nawet z zaangażowaniem TRAIL, prokaspaza-8 w DISC nie może być aktywowana w pełni i w związku z tym nie wystąpi apoptoza z udziałem TRAIL.
 Niska aktywność wewnątrzkomórkowego PKCK2 (1) lub wysoka aktywność wewnątrzkomórkowego PKCK2 jest obniżana przez specyficzny inhibitor (1’), defosforylowane monomery prokaspazy-2. Prokaspaza-2 jest następnie aktywowana w wyniku jej dimeryzacji (2), a zaktywowana kaspaza-2 tnie monomer prokaspazy-8 pomiędzy większą i mniejszą podjednostką (3). W takiej sytuacji dochodzi do ‘wzbudzenia’ apoptozy zależnej od TRAIL w komórkach nowotoworowych. Jeżeli TRAIL nie jest połączony z receptorami TRAIL-śmierć, cięta prokaspaza-8 jest kierowana do proteasomu celem degradacji (4). W przypadku przeciwnym TRAIL jest wiązany do swego receptora, przecięta prokaspaza-8 jest rekrutowana przez receptory śmierci TRAIL, czego wynikiem jest utworzenie DISC (4’). Drugie cięcie między prodomeną i podjednostką większą może być wykonane efektywnie w wyniku dimeryzacji ciętych prokaspaz-8, która zachodzi z udziałem DISC (5), co prowadzi do aktywacji prokaspazy-8 (6), a następnie apoptozy za pośrednictwem TRAIL (7). (Prawy schemat) Gdy wewnątrzkomórkowa aktywność PKCK2 jest wysoka prokaspaza-2 nie może być aktywowana; a zatem prokaspaza-8 nie może być przekształcana. Nawet z zaangażowaniem TRAIL, prokaspaza-8 w DISC nie może być aktywowana w pełni i w związku z tym nie wystąpi apoptoza z udziałem TRAIL.")
80
Śmierć kaspazo-zależna Śmierć kaspazo-niezależna
Śmierć z aktywacją kaspaz Śmierć z aktywacją kaspaz Śmierć bez aktywacji kaspaz Przeżycie Apoptoza Nekroza Śmierć apoptozo-podobna Śmierć autofagowa Apoptyczna śmierć komórki Niapoptyczna śmierć komórki Wpływ blokerów kaspaz Wyznaczone ścieżki Związki między kaspazo-zależnością i morfotypem śmierci komórki

81
M U T A C J E i ZaBU rzeNia PrZez REGULATORÓW A P O P T O Z Y CYKLU
KOMÓRKOWEGO i

82
MUTACJE NIEAKTYWNE LIGANDY
pro-kaspaza-8 Domena zewnątrz-komórkowa FasL Fas/CD95 Błona komórkowa FADD MUTACJE NIEAKTYWNE RECEPTORY Jądro komórkowe Bcl2 MUTACJE NIEAKTYWNE KINAZY Kaspazy wykonawcze & inne substraty kaspaza 3 cytochrom c kaspaza-9/cytochrom c mitochondria p15tBid ???PRZEMIANA NOWOTWOROWA??? = MUTACJE NIEAKTYWNA KASPAZA 8

83
(osiem białek znanych do 2001)
Blokują apoptozę (osiem białek znanych do 2001)
")
84
MUTACJE NIEAKTYWNE LIGANDY
pro-kaspaza-8 Domena zewnątrz-komórkowa FasL Fas/CD95 Błona komórkowa FADD Jądro komórkowe Bcl2 MUTACJE - NIEAKTYWNA KASPAZA 8 MUTACJE - NIEAKTYWNE RECEPTORY p53 Bax MUTACJE - NIEAKTYWNE KINAZY Kaspazy wykonawcze & inne substraty kaspaza 3 cytochrom c kaspaz-9/cytochrom c p15tBid mitochondria

85
NAPRAWA LUB APOPTOZA p53 p21WAF1/CIP CYKLINA Cdk CYKLINA : Cdk ATP
pRB : E2F ADP pRB : E2F ppRB : E2F E2F FAZA G1 FAZA S FAZA G1 FAZA S NAPRAWA LUB APOPTOZA

86
(czternaście białek znanych do 2001)
Wzbudzają apoptozę (czternaście białek znanych do 2001)
")
87
!!!PRZEMIANA NOWOTWOROWA!!!
pro-kaspaza-8 Domena zewnątrz-komórkowa FasL Fas/CD95 Błona komórkowa FADD Jądro komórkowe Bcl2 MUTACJE NIEAKTYWNA KASPAZA 8 mitochondria p53 Bax MUTACJE MUTACJE MUTACJE !!!PRZEMIANA NOWOTWOROWA!!!

88
Complex

89
ZEWNĄTRZ CYTOPLAZMA Complex

90
BRAK REKRUTACJI DO COMPLEKSU ŚMIERCI BRAK AKTYWNOŚCI PROTEO- LITYCZNEJ
(Wyniki mutacji) BRAK REKRUTACJI DO COMPLEKSU ŚMIERCI BRAK AKTYWNOŚCI PROTEO- LITYCZNEJ
 BRAK. REKRUTACJI. DO. COMPLEKSU. ŚMIERCI. BRAK. AKTYWNOŚCI. PROTEO- LITYCZNEJ.")
91
Mutacje czynników wzbudzających apoptozę
***

92
NIEKONTROLOWANE PODZIAŁY GROMADZENIE MUTACJI
p21WAF1/CIP CYKLINA : Cdk pRB : E2F ppRB : E2F E2F ATP ADP FAZA G1 FAZA S CYKLINA Cdk NAPRAWA LUB APOPTOZA MUTACJE NIEKONTROLOWANE PODZIAŁY GROMADZENIE MUTACJI

93
NIEKONTROLOWANE PODZIAŁY GROMADZENIE MUTACJI
p21WAF1/CIP CYKLINA : Cdk pRB : E2F ppRB : E2F E2F ATP ADP FAZA G1 FAZA S CYKLINA Cdk NAPRAWA LUB APOPTOZA MUTACJE NIEKONTROLOWANE PODZIAŁY GROMADZENIE MUTACJI

94
NIEKONTROLOWANE PODZIAŁY GROMADZENIE MUTACJI
p21WAF1/CIP CYKLINA : Cdk pRB : E2F ppRB : E2F E2F ATP ADP FAZA G1 FAZA S CYKLINA Cdk NAPRAWA LUB APOPTOZA MUTACJE NIEKONTROLOWANE PODZIAŁY GROMADZENIE MUTACJI

95
ZABURZENIA APOPTOZY 1. MUTACJE CZYNNIKÓW ZAANGAŻOWANYCH W APOPTOZĘ
A/ RECEPTORY np. dla TNF lub Fas B/ LIGANDY C/ PROTEINAZY D/ TELOMERAZA E/ INNE ENZYMY (np.: DNA-azy, itp.) 2. MUTACJE CZYNNIKÓW CYKLU KOMÓRKOWEGO A/ KINAZY ZALEŻNE OD CYKLIN B/ CYKLINY 3. MUTACJE REGULATORÓW CYKLU KOMÓRKOWEGO A/ p53 B/ pRB C/ p21/WAF/CIP D/ BRCA
 2. MUTACJE CZYNNIKÓW CYKLU KOMÓRKOWEGO. A/ KINAZY ZALEŻNE OD CYKLIN. B/ CYKLINY. 3. MUTACJE REGULATORÓW CYKLU KOMÓRKOWEGO. A/ p53. B/ pRB. C/ p21/WAF/CIP. D/ BRCA.")
96
REMEDIUM???

97
Wymuszenie blokady cyklu komórkowego i aktywacja apoptozy

98
NOWE PODEJŚCIE DO LECZENIA NOWOTWORÓW
TECHNOLOGIA SAANDs (Selective Apoptotic Antineoplastic Drugs) NOWE PODEJŚCIE DO LECZENIA NOWOTWORÓW Opracowane przez firmę biotechnologiczną Cell Pathways, Inc. Aptosyn™ (exisulind). Jest to kandydat nowej generacji leków. Wywołuje apoptozę wybiórczo w komórkach przed- i nowotworowych, przez co tylko komórki nowotworowe są zmuszane do samounicestwienia. W minimalnym stopniu uszkadza komórki normalne.
 NOWE PODEJŚCIE DO LECZENIA NOWOTWORÓW. Opracowane przez firmę biotechnologiczną Cell Pathways, Inc. Aptosyn™ (exisulind). Jest to kandydat nowej generacji leków. Wywołuje apoptozę wybiórczo w komórkach przed- i nowotworowych, przez co tylko komórki nowotworowe są zmuszane do samounicestwienia. W minimalnym stopniu uszkadza komórki normalne.")
99
ZAŁOŻENIA TECHNOLOGII SAANDs
NOWE LEKI MUSZĄ: Wzbudzać zaprogramowaną śmierć komórki (apoptozę) wyłącznie w komórkach przed- i nowotworowych. Wykazywać aktywność w szerokim spektrum linii komórek nowotworowych (wrażliwość na Aptosyn™ testowano dla ponad 50 linii). Działać synergicznie lub kumulująco z lekami chemioterapii konwencjonalnej. Posiadać mechanizm działania niezależny od dotychczas znanych szlaków regulujących apoptozę.
 wyłącznie w komórkach przed- i nowotworowych. Wykazywać aktywność w szerokim spektrum linii komórek nowotworowych (wrażliwość na Aptosyn™ testowano dla ponad 50 linii). Działać synergicznie lub kumulująco z lekami chemioterapii konwencjonalnej. Posiadać mechanizm działania niezależny od dotychczas znanych szlaków regulujących apoptozę.")
100
komórek raka odbytu człowieka (HT-29)
HODOWLA komórek raka odbytu człowieka (HT-29) KONTROLA Komórka rakowa oporna Komórka rakowa nekrotyczna Komórki rakowe w obecności Aptosyn™ Komórka rakowa podlegająca Apoptozie
 KONTROLA. Komórka rakowa. oporna. Komórka rakowa. nekrotyczna. Komórki rakowe. w obecności Aptosyn™ Komórka rakowa. podlegająca. Apoptozie.")
101
komórek raka prostaty człowieka (PC-3)
HODOWLA komórek raka prostaty człowieka (PC-3) Komórka rakowa nie podlegająca apoptozie KONTROLA Komórki rakowe w obecności Aptosyn™ Komórka rakowa podlegająca apoptozie
 Komórka. rakowa. nie. podlegająca. apoptozie. KONTROLA. Komórki rakowe. w obecności Aptosyn™ Komórka. rakowa. podlegająca. apoptozie.")
102
EFEKTY TECHNOLOGII SAANDs
Blokuje cykliczny GMP-PDE. Aktywuje kinazę białkową G Może być stosowany szeroko w chemo-prewencji przy leczeniu zaawansowa-nych nowotoworów

103
EFEKTY TECHNOLOGII SAANDs (c.d. 2)
Nie blokuje COX I lub II i dlatego nie powoduje zagrożeń toksycznych, żołądkowo-jelitowego i nerkowego, typowych dla leków NSAIDs. Nie posiada działań ubocznych charakterystycz-nych dla terapii hormonalnych i chemioterapii. Jest stosowany doustnie

104
Konwencjonalna chemioterapia
PORÓWNANIE Konwencjonalna chemioterapia i radioterapia wzbudzają nekrozę i apoptozę we wszystkich proliferujących komórkach. SAANDs wzbudza apoptozę wyłącznie w komórkach zmienionych nowotworowo (neoplastycznych) Istniejące leki chemioterapii oraz radioterpia wzbudzają apoptozę w szybko dzielących się komórkach bez rozróżnienia między komórkami neoplastycznymi i normalnymi. Śmierć normalnych komórek prowokuje liczne nieporządane skutki. Aptosyn™ i inne leki grupy SAANDs, takie jak CP-461 i CP-248 wzbudzają apoptozę wyłącznie w komórkach nowotworowych, jak to pokazano w badaniach ponad 50 różnych linii komórek nowotworowych, nie wywołując śmierci w komórkach sąsiednich w leczonej tkance.
 Istniejące leki chemioterapii oraz radioterpia wzbudzają apoptozę w szybko dzielących się komórkach bez rozróżnienia między komórkami neoplastycznymi i normalnymi. Śmierć normalnych komórek prowokuje liczne nieporządane skutki. Aptosyn™ i inne leki grupy SAANDs, takie jak CP-461 i CP-248 wzbudzają apoptozę wyłącznie w komórkach nowotworowych, jak to pokazano w badaniach ponad 50 różnych linii komórek nowotworowych, nie wywołując śmierci w komórkach sąsiednich w leczonej tkance.")
105
MECHANIZM DZIAŁANIA APTOSYNTM
APOPTOZA Aktywna Kaspaza Sygnał apoptotyczny GTP Cyklaza guanylowa cGMP-PDE Aktywny GMP Cykliczny GMP MECHANIZM DZIAŁANIA APTOSYNTM Aktywna Kinaza białkowa G b-katenina Kinaza białkowa G Aktywna Kaspaza

106
APTOSYNTM blokuje cGMP-PDE
Aktywna Kinaza białkowa G b-katenina APOPTOZA Sygnał apoptotyczny GTP Cyklaza guanylowa cGMP-PDE Cykliczny GMP Kaspaza APTOSYNTM blokuje cGMP-PDE

107
regulatorów apoptozy takich jak: nie blokuje aktywności
APTOSYN™ Działa za pośrednictwem regulatorów apoptozy takich jak: p53, Bcl2 i Bax oraz nie blokuje aktywności COX I i II.

108
STOSOWANIE APTOSYNUTM u PACJENTA Z FAP/APC i GS
(Familial Adenomatous Polyposis Coli) Polipy po stoswaniu AptosynuTM przez 6 miesięcy Polipy nieleczone Polipy po stoswaniu AptosynuTM przez 44 miesiące
 Polipy po stoswaniu. AptosynuTM. przez 6 miesięcy. Polipy nieleczone. Polipy po stoswaniu. AptosynuTM. przez 44 miesiące.")
109
pozwala uniknąć efektów ubocznych
SAANDs pozwala uniknąć efektów ubocznych Radioterapia i liczne standardowe chemioterapeutyki takie jak np.: 5-fluorouracyl egzekwują kontrolę nowotworów przez specyficzne i niespecyficzne szlaki ragulatorowe. Aktywność apoptotyczna wywołana chemioterapeutykami i promieniowaniem jest wtórnym wynikiem ich stosowania wywołującego uszkodzenia w komórce.

110
ZALETY SAANDs Leki SAANDs działają poprzez korektę uszkodzenia w głównym szlaku regulującym apoptozę. Odtwarzając przekazywanie sygnału szlakiem apoptotycznym, SAANDs stają się naczelnym aktywatorem apo-ptozy o najwyższej specyficzności w komórkach przeznaczonych de facto do usunięcia.







 ZALECANE PODRĘCZNIKI (do przygotowania do ćwiczeń i uzupełnienia.>")













