Pobierz prezentację
This is a modal window.
1
Nowotwory mieloproliferacyjne Przewlekłe zespoły mieloproliferacyjne

2
Nowotwory mieloproliferacyjne
Definicja: klonalne choroby będące następstwem nowotworowej transformacji krwiotwórczej komórki macierzystej, z nadmierną proliferacją jednej lub więcej linii mieloidalnych w szpiku Cechy wspólne zwiększenie liczby granulocytów i/lub krwinek czerwonych i/lub płytek we krwi obwodowej Splenomegalia: sekwestracji nadmiaru komórek krwi, pozaszpikowe krwiotworzenie lub nacieki białaczkowe progresja w zwłóknienie szpiku lub transformacja w ostrą białaczkę

3
Nowotwory mieloproliferacyjne
Przewlekła białaczka szpikowa Czerwienica prawdziwa Nadpłytkowość samoistna Pierwotne zwłóknienie szpiku Przewlekła białaczka eozynofilowa Przewlekła białaczka neutrofilowa Mastocytoza Mieloproliferacja niesklasyfikowana WHO 2008

4
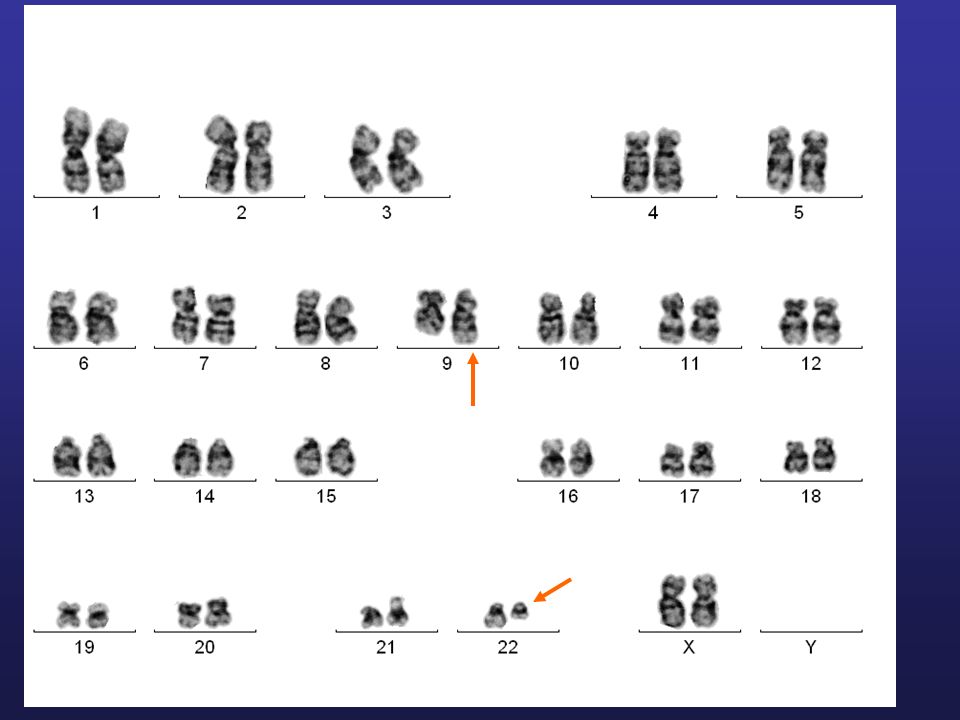
Przewlekła białaczka szpikowa (pbsz)
Choroba klonalna komórki macierzystej szpiku związana z występowaniem chromosomu Philadelphia i genu fuzyjnego BCR/ABL Nieprawidłowy gen fuzyjny znajdowany jest we wszystkich liniach mieloidalnych, a także w części komórek limfoidalnych Chromosom Philadelfia - t(9;22) - stanowi stałą aberrację cytogenetyczną w pbsz Translokacja (9;22) prowadzi do przeniesienia z chromosomu 9 genu ABL w rejon genu BCR na chromosomie 22; gen fuzyjny BCR/ABL koduje kinazę tyrozyny o nieprawidłowej aktywności, co prowadzi do wzmożonej proliferacji nowotworowej komórki macierzystej zahamowania apoptozy zaburzonej funkcji integryn, powodując upośledzenie adhezji komórek białaczkowych do podścieliska szpiku
 - stanowi stałą aberrację cytogenetyczną w pbsz. Translokacja (9;22) prowadzi do przeniesienia z chromosomu 9 genu ABL w rejon genu BCR na chromosomie 22; gen fuzyjny BCR/ABL koduje kinazę tyrozyny o nieprawidłowej aktywności, co prowadzi do. wzmożonej proliferacji nowotworowej komórki macierzystej. zahamowania apoptozy. zaburzonej funkcji integryn, powodując upośledzenie adhezji komórek białaczkowych do podścieliska szpiku.")
5
Epidemiologia Częstość występowania 1-2 osób/ populacji rocznie Częściej chorują mężczyźni niż kobiety (1,3:1) Mediana wieku w chwili rozpoznania 60 lat

6
Przebieg i objawy kliniczne
Faza przewlekła bezobjawowo u około 20-40% chorych (rozpoznanie przypadkowe) osłabienie, spadek masy ciała, poty nocne, objawy związane ze splenomegalią, zaburzenia widzenia, bóle głowy Faza akceleracji stany gorączkowe, bóle kostne, osłabienie, skaza krwotoczna, bóle brzucha związane z olbrzymich rozmiarów splenomegalią Faza blastyczna objawy jak w ostrej białaczce
 osłabienie, spadek masy ciała, poty nocne, objawy związane ze splenomegalią, zaburzenia widzenia, bóle głowy. Faza akceleracji. stany gorączkowe, bóle kostne, osłabienie, skaza krwotoczna, bóle brzucha związane z olbrzymich rozmiarów splenomegalią. Faza blastyczna. objawy jak w ostrej białaczce.")
7
Diagnostyka Morfologia Rozmaz krwi obwodowej FAG (0-30 score)
leukocytoza niedokrwistość (nie zawsze) nadpłytkowość (często) Rozmaz krwi obwodowej szereg rozwojowy granulocytów (odmłodzenie do mieloblasta) eozynofilia bazofilia erytroblasty FAG (0-30 score) Kwas moczowy LDH USG jamy brzusznej - splenomegalia
 nadpłytkowość (często) Rozmaz krwi obwodowej. szereg rozwojowy granulocytów (odmłodzenie do mieloblasta) eozynofilia. bazofilia. erytroblasty. FAG (0-30 score) Kwas moczowy. LDH. USG jamy brzusznej - splenomegalia.")
8
Diagnostyka Badanie cytogenetyczne szpiku metodą klasyczną i/lub FISH i/lub badanie molekularne chromosom Philadelphia rearanżacja bcr/abl Biopsja aspiracyjna szpiku hiperplazja, z dominacją linii granulocytarnej Trepanobiopsja szpiku nasilone włóknienie kolagenowe i retikulinowe

9
Diagnostyka Badania cytogenetyczne Metody Klasyczna metoda prążkowa
FISH Wskazania Diagnostyka Monitorowanie leczenia

10
Diagnostyka Badanie molekularne Metody Jakościowa Ilościowa
Badanie mutacji Wskazania Diagnostyka Monitorowanie leczenia

11
Chromosom Filadelfia

13
FISH - t(9;22)(q34;q11) Dual Fusion Dual Color, Dual Fusion BCR/ABL
Komórka prawidłowa Dual Fusion Komórka nieprawidłowa z t(9;22), sonda Dual Color, Dual Fusion
, sonda Dual Color, Dual Fusion.")
14
Diagnostyka Kryteria fazy akceleracji Kryteria kryzy blastycznej
blasty we krwi obwodowej lub szpiku 10-19% (-29% wg ELN) bazofilia krwi 20% małopłytkowość < 100 G/l (nie związana z leczeniem) nadpłytkowość > 1000 G/l klonalna ewolucja cytogenetyczna (dodatkowe aberracje chromosomowe) powiększenie śledziony lub wzrost leukocytozy nie reagujący na terapię Kryteria kryzy blastycznej odsetek blastów ≥ 20% (30% wg ELN) pozaszpikowe nacieki białaczkowe WHO
 bazofilia krwi 20% małopłytkowość < 100 G/l (nie związana z leczeniem) nadpłytkowość > 1000 G/l. klonalna ewolucja cytogenetyczna (dodatkowe aberracje chromosomowe) powiększenie śledziony lub wzrost leukocytozy nie reagujący na terapię. Kryteria kryzy blastycznej. odsetek blastów ≥ 20% (30% wg ELN) pozaszpikowe nacieki białaczkowe. WHO.")
15
Modele rokownicze w pbsz
Modele rokownicze służą do przewidywania czasu do wystąpienia przełomu blastycznego, pomocne w wyborze metody leczenia u chorego na pbsz Powszechnie uznane modele rokownicze (wskaźnik Sokala, wskaźnik Hasforda, Euro, Eutos) uwzględniają następujące parametry: wiek chorego wielkość śledziony odsetek blastów w krwi obwodowej liczbę płytek krwi odsetek bazofili i eozynofili w krwi
 uwzględniają następujące parametry: wiek chorego. wielkość śledziony. odsetek blastów w krwi obwodowej. liczbę płytek krwi. odsetek bazofili i eozynofili w krwi.")
16
Przewlekła białaczka szpikowa – rekomendacje European LeukemiaNet 2006, 2009, 2013
Diagnostyka Leczenie Terapia pierwszej linii Terapia drugiej linii Odpowiedź na leczenie – definicje Zasady monitorowania leczenia Baccarani et al. Blood 2006, Blood 2013 Baccarani et al. JCO 2009

17
Przewlekła białaczka szpikowa – rekomendacje European LeukemiaNet 2006, 2009, 2013
Leczenie Terapia pierwszej linii Imatinib (Glivec) 400 mg/dobę Dasatinib (Sprycel) 100 mg/d Nilotinib (Tasigna) 600 mg/d Terapia drugiej linii Inhibitory kinazy tyrozynowej pierwszej lub drugiej generacji Bosutinib Ponatinib alloHSCT Terapia eksperymentalna Baccarani et al. Blood 2006, Blood 2013 Baccarani et al. JCO 2009
 400 mg/dobę. Dasatinib (Sprycel) 100 mg/d. Nilotinib (Tasigna) 600 mg/d. Terapia drugiej linii. Inhibitory kinazy tyrozynowej pierwszej lub drugiej generacji. Bosutinib. Ponatinib. alloHSCT. Terapia eksperymentalna. Baccarani et al. Blood 2006, Blood Baccarani et al. JCO")
18
Leczenie Inhibitory kinazy tyrozynowej (TKI) - leczenie umożliwia uzyskanie remisji hematologicznej u 95-99% chorych, większej odpowiedzi cytogenetycznej u 85% chorych, w tym pełnej odpowiedzi u 87% chorych (MMR 50%) Imatinib (Glivec), Dazatynib (Sprycel), Nilotinib (Tasigna) Kolejne linie: ponatinib (45mg), bosutinib (500mg) Przeszczepienie allogenicznych komórek hematopoetycznych wieloletnie przeżycie osiąga około 50% chorych ryzyko zgonu z powodu powikłań związanych z leczeniem wynosi ok. 10% ryzyko wznowy choroby wynosi 10-20% Leczenie cytoredukcyjne - Hydroksykarbamid, Busulfan leczenie nie przedłuża życia chorych na pbsz, nie powoduje przedłużenia fazy przewlekłej choroby, ale poprawia komfort życia Interferon alfa leczenie umożliwia uzyskanie remisji hematologicznej u 50-80% chorych, większej odpowiedzi cytogenetycznej u 30% oraz pełnej remisji cytogenetycznej u < 10% chorych Przeszczepienie autologicznych komórek hematopoetycznych przedłuża życie, ale nie można wyleczyć choroby obecnie prowadzone jedynie w ramach programów badawczych
 - leczenie umożliwia uzyskanie remisji hematologicznej u 95-99% chorych, większej odpowiedzi cytogenetycznej u 85% chorych, w tym pełnej odpowiedzi u 87% chorych (MMR 50%) Imatinib (Glivec), Dazatynib (Sprycel), Nilotinib (Tasigna) Kolejne linie: ponatinib (45mg), bosutinib (500mg) Przeszczepienie allogenicznych komórek hematopoetycznych. wieloletnie przeżycie osiąga około 50% chorych. ryzyko zgonu z powodu powikłań związanych z leczeniem wynosi ok. 10% ryzyko wznowy choroby wynosi 10-20% Leczenie cytoredukcyjne - Hydroksykarbamid, Busulfan. leczenie nie przedłuża życia chorych na pbsz, nie powoduje przedłużenia fazy przewlekłej choroby, ale poprawia komfort życia. Interferon alfa. leczenie umożliwia uzyskanie remisji hematologicznej u 50-80% chorych, większej odpowiedzi cytogenetycznej u 30% oraz pełnej remisji cytogenetycznej u < 10% chorych. Przeszczepienie autologicznych komórek hematopoetycznych. przedłuża życie, ale nie można wyleczyć choroby. obecnie prowadzone jedynie w ramach programów badawczych.")
19
IRIS International Randomized Study of IFN+AraC vs STI571
2003 rok – okres obserwacji 19 miesięcy CHR 96% MCyR 87% CCyR 76% 2006 rok – okres obserwacji 5 lat CHR 98% CCyR 87% OS 89,5% Progresja do AP/BC 7% Wznowa % O’Brien et al.; N Engl J Med. 2003 Druker et al.; N Engl J Med. 2006

20
Prawdopodobieństwo przeżycia oraz śmiertelność związana z transplantacją po alloHSCT u chorych na PBSz w zależności od ryzyka transplantacyjnego Gratwohl et al.; Lancet 1998

21
Przewlekła białaczka szpikowa
AlloSCT w CML. Gratwohl et al. BMT 2011

22
Przewlekła białaczka szpikowa
AlloSCT w CML. TRM 8%. Saussele et al. Blood 2010

23
Leczenie Powodzenie leczenia zależy od właściwego monitorowania choroby na poziomie hematologicznym cytogenetycznym molekularnym

24
Kryteria odpowiedzi hematologicznej
Ustąpienie objawów ogólnych Ustąpienie splenomegalii Morfologia: normalizacja Rozmaz krwi obwodowej: normalizacja

25
Kryteria odpowiedzi cytogenetycznej
Odpowiedź cytogenetyczna Odsetek komórek Ph+ w szpiku całkowita większa 1 - 35 mniejsza brak > 95

26
Kryteria odpowiedzi molekularnej
Całkowita odpowiedź molekularna Brak transkryptu bcr/abl w reakcji jakościowego PCR Większa odpowiedź molekularna 3-log redukcja transkryptu bcr/abl w stosunku do wyjściowego w badaniu ilościowym PCR

27
Przewlekła białaczka szpikowa – rekomendacje European LeukemiaNet 2013
Baccarani et al. Blood 2013

28
Czerwienica prawdziwa
Definicja: nowotwór mieloproliferacyjny, wynikający z klonalnej proliferacji krwiotwórczej komórki macierzystej, charakteryzujący się zwiększoną produkcją komórek czerwonokrwinkowych, niezależną od fizjologicznych mechanizmów regulujących erytropoezę

29
Epidemiologia Częstość występowania 1 / 100 000 populacji / rok
Częściej chorują mężczyźni niż kobiety (1-2:1) Średni wiek w chwili rozpoznania lat
 Średni wiek w chwili rozpoznania lat.")
30
Czerwienica prawdziwa- kryteria rozpoznania
Kryteria większe Hb > 18,5 g/dl (M) Hb > 16,5 g/dl (K) mutacja JAK2 lub defekt molekularny o podobnych konsekwencjach Kryteria mniejsze szpik bogatokomórkowy w odniesieniu do linii erytro-, granulo- i megakariocytowej (panmielosis) erytropetyna poniżej normy endogenny wzrost kolonii erytroidalnych
 Hb > 16,5 g/dl (K) mutacja JAK2 lub defekt molekularny o podobnych konsekwencjach. Kryteria mniejsze. szpik bogatokomórkowy w odniesieniu do linii erytro-, granulo- i megakariocytowej (panmielosis) erytropetyna poniżej normy. endogenny wzrost kolonii erytroidalnych.")
31
Przyczyny wtórnej nadkrwistości
Hipoksemia: sinicze wady serca przewlekłe choroby układu oddechowego: POCHP, zespół bezdechu sennego przebywanie na dużej wysokości Choroby nerek: torbiele nerek, wodonercze, stan po przeszczepieniu nerki Nowotwory wątroby, móżdżku, gruczolaki nadnercza, guz chromochłonny nadnerczy, mięśniaki macicy Zaburzenia endokrynologiczne: hiperaldosteronizmem

32
Nadkrwistość względna (rzekoma)
odwodnienie zwiększona utrata białka (oparzenia, enteropatia) nadmierna otyłość nadmierne spożycie alkoholu zespół Geisbocka (zaburzenie regulacji objętości osocza) - mężczyźni skłonni do stresów, z nadciśnieniem tętniczym hipercholesterolemią, hipelipidemią
 nadmierna otyłość. nadmierne spożycie alkoholu. zespół Geisbocka (zaburzenie regulacji objętości osocza) - mężczyźni skłonni do stresów, z nadciśnieniem tętniczym hipercholesterolemią, hipelipidemią.")
33
Objawy kliniczne Bóle, zawroty głowy Uporczywy świąd skóry
Erytromelalgia Zwiększona potliwość, osłabienie Powikłania zakrzepowo-zatorowe (20-30% chorych) zakrzepica tętnicza naczyń mózgowych i naczyń wieńcowych zakrzepica żylna- nietypowe lokalizacja: zakrzepica żył krezkowych, żyły śledzionowej, wrotnej Powikłania krwotoczne (20% chorych)
 zakrzepica tętnicza naczyń mózgowych i naczyń wieńcowych. zakrzepica żylna- nietypowe lokalizacja: zakrzepica żył krezkowych, żyły śledzionowej, wrotnej. Powikłania krwotoczne (20% chorych)")
34
Rokowanie Średni czas życia chorych 15-20 lat
Większość chorych umiera z powodu powikłań zakrzepowo-zatorowych lub krwotocznych U około 20% chorych rozwija się zespół mielodysplastyczny lub ostra białaczka U części chorych choroba ewoluuje w kierunku mielofibrozy

35
Leczenie Krwioupusty ml powtarzane do czasu normalizacji wartości hematokrytu Leki mielosupresyjne Hydroksykarbamid mg dziennie Anagrelid Interferon (wybrane sytuacje kliniczne np. ciąża) Pipobroman Busulfan, Melfalan, Chlorambucyl Leczenie antyagregacyjne Inne leki i metody Fosfor radioaktywny 32P (USA) Inhibitory JAK2
 Pipobroman. Busulfan, Melfalan, Chlorambucyl. Leczenie antyagregacyjne. Inne leki i metody. Fosfor radioaktywny 32P (USA) Inhibitory JAK2.")
36
Nadpłytkowość samoistna
Choroba z grupy nowotworów mieloproliferacyjnych cechująca się zwiększoną liczbą płytek krwi, wywołaną klonalnym rozrostem megakariocytów Częstość występowania 5-7 osób / populacji Częściej chorują mężczyźni niż kobiety (1,2:1) Dwa szczyty zachorowań- młode kobiety i u osób między 50 a 70 rokiem życia
 Dwa szczyty zachorowań- młode kobiety i u osób między 50 a 70 rokiem życia.")
37
Objawy kliniczne Bezobjawowo u części chorych
Powikłania zakrzepowo-zatorowe u 70% chorych (zakrzepica żylna i tętnicza o różnej lokalizacji) Objawy skazy krwotocznej u 10-15% chorych Erytromelalgia - u 85% chorych, może doprowadzić do martwicy palców
 Objawy skazy krwotocznej u 10-15% chorych. Erytromelalgia - u 85% chorych, może doprowadzić do martwicy palców.")
38
Kryteria rozpoznania Liczba płytek krwi powyżej 450 G/L (utrzymująca się) Proliferacja linii megakariocytowej w szpiku Klonalne zaburzenia molekularne: mutacja JAK2 Kryteria z wykluczenia: Czerwienica prawdziwa Przewlekła białaczka szpikowa Mielofibroza Wykluczenie przyczyn wtórnej nadpłytkowości Brak cech mielodysplazji (ocena morfologiczna, badania cytogenetyczne)
")
39
Przyczyny wtórnej nadpłytkowości
ostra utrata krwi niedobór żelaza stan po splenektomii nowotwory (płuc, trzustki) przewlekłe choroby zapalne i infekcyjne alkoholizm polekowo stali dawcy krwi
 przewlekłe choroby zapalne i infekcyjne. alkoholizm. polekowo. stali dawcy krwi.")
40
Leczenie Wszyscy chorzy
leczenie odwracalnych chorób i stanów sprzyjających powikłaniom naczyniowym (nadciśnienie tętnicze, palenie papierosów, otyłość, hipercholestrolemia) Chorzy z wysokim ryzykiem powikłań naczyniowych (wiek > 60 lat, liczba płytek powyżej 1500 G/L, epizod zakrzepowy w wywiadzie) Hydrokskarbamid + małe dawki aspiryny (100 mg/dobę) Anagrelid lub interferon: leczenie drugiej linii Chorzy z ryzykiem pośrednim powikłań naczyniowych (wiek lat, brak czynników wysokiego ryzyka) małe dawki aspiryny +/- hydroksyurea (u wybranych chorych) Chorzy z małym ryzykiem powikłań naczyniowych (< 40 lat) małe dawki aspiryny
 Chorzy z wysokim ryzykiem powikłań naczyniowych (wiek > 60 lat, liczba płytek powyżej 1500 G/L, epizod zakrzepowy w wywiadzie) Hydrokskarbamid + małe dawki aspiryny (100 mg/dobę) Anagrelid lub interferon: leczenie drugiej linii. Chorzy z ryzykiem pośrednim powikłań naczyniowych (wiek lat, brak czynników wysokiego ryzyka) małe dawki aspiryny +/- hydroksyurea (u wybranych chorych) Chorzy z małym ryzykiem powikłań naczyniowych (< 40 lat) małe dawki aspiryny.")
41
Pierwotne zwłóknienie szpiku
Klonalna proliferacja megakariocytów i granulocytów w szpiku z wtórnym postępującym rozplemem włókien łącznotkankowych w jamach szpikowych kości i występowaniem ognisk metaplazji pozaszpikowej Choroba rzadko występująca 5 osób / populacji Średnia wieku chorych 65 lat

42
Objawy kliniczne 30% - bezobjawowa objawowa znaczna splenomegalia
niedokrwistość bóle kostne i tkliwość opukowa kości osłabienie, spadek wagi, nocne poty, stany gorączkowe powikłania zakrzepowo-zatorowe krwawienia

43
Badania laboratoryjne
Morfologia niedokrwistość, leukocytoza (faza przedfibrotyczna) trombocytoza (faza przedfibrotyczna) Rozmaz krwi: odczyn leukoerytroblastyczny, krwinki czerwone w kształcie łzy, erytroblasty, poikilocytoza Kwas moczowy LDH Biopsja aspiracyjna szpiku - „pusta” Trepanobiopsja - włóknienie retikulinowe/kolagenowe
 trombocytoza (faza przedfibrotyczna) Rozmaz krwi: odczyn leukoerytroblastyczny, krwinki czerwone w kształcie łzy, erytroblasty, poikilocytoza. Kwas moczowy. LDH. Biopsja aspiracyjna szpiku - „pusta Trepanobiopsja - włóknienie retikulinowe/kolagenowe.")
44
Mielofibroza – kryteria diagnostyczne
Kryteria większe Proliferacja w linii megakariocytowej z atypią oraz włóknienie retikulinowe lub kolagenowe w szpiku Wykluczenie czerwienicy prawdziwej, pbsz, mds i innych nowotworów Mutacja JAK2 Wykluczenie reaktywnego zwłóknienia szpiku Kryteria mniejsze Leukoerytroblastoza krwi Anemia Splenomegalia Wzrost LDH

45
Niekorzystne czynniki rokownicze
Wiek > 65 lat Niedokrwistość Hb < 10g/dl Leukocytoza krwi > 25 k/ul Zwiększony odsetek blastów we krwi >1% Objawy ogólne (gorączka, spadek masy ciała) – przynajmniej jeden
 – przynajmniej jeden.")
46
Leczenie Leki mielosupresyjne Hydroksykarbamid Busulfan
Leki hormonalne androgeny kortykosteroidy Napromienianie śledziony Splenektomia Transplantacja allogenicznych komórek hematopoetycznych Interferon Inhibitory JAK2 Ruxolitinib

















 – współczynnik powstały przez podzielenie masy.>")

>")

