Hemostaza Funkcją hemostazy jest: ochrona przed utratą krwi zatrzymanie krwawienia zapobieżenie zakrzepicy
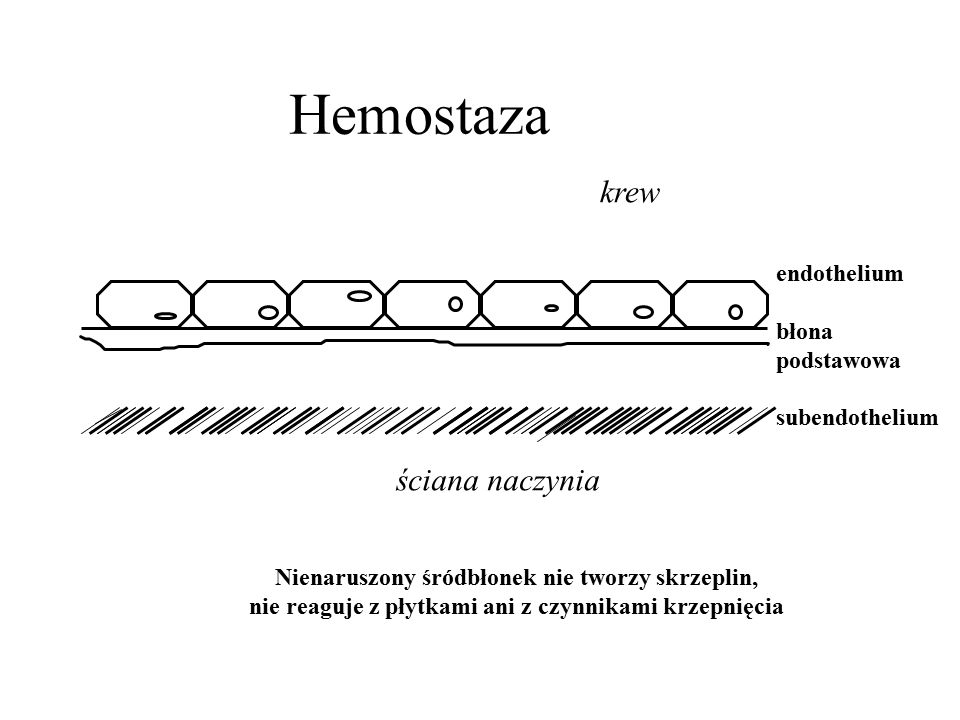
Hemostaza krew ściana naczynia endothelium błona podstawowa subendothelium ściana naczynia Nienaruszony śródbłonek nie tworzy skrzeplin, nie reaguje z płytkami ani z czynnikami krzepnięcia
Hemostaza podstawowa krew uszkodzenie ściany naczynia płytki czynniki endothelium błona podstawowa subendothelium uszkodzenie ściany naczynia Gdy naczynie jest uszkodzone uwalniają się czynniki tkankowe zaś warstwa podśródbłonkowa wyeksponowana jest na działanie płytek i czynników krzepnięcia
Hemostaza Hemostaza jest procesem trójfazowym składającym się z: hemostazy pierwotnej krzepnięcia fibrynolizy
Konsolidacja czopu płytkowego Krzepnięcie Powstawanie skrzepu Konsolidacja czopu płytkowego za pomocą włóknika
Fibrynoliza fibrynoliza rozpuszczenie skrzepu ochrona przed zakrzepicą
Równowaga w układzie krzepnięcia czynniki krzepnięcia fosfolipidy Ca ++ tPA uPA + + FIBRYNA krzepnięcie trombina fibrynoliza plazmina _ _ AT III białko C białko S PAI-1 antyplazmina
bezjądrzaste elementy morfotyczne krwi PŁYTKI KRWI = TROMBOCYTY – PLT bezjądrzaste elementy morfotyczne krwi powstają w szpiku w procesie trombopoezy z megakariocytów przy udziale trombopoetyny, Il-3, 11 czas przeżycia płytek – 8-12 dni (rozpad w ukł. s-ś śledziony i wątroby) wartości prawidłowe PLT PLT = 150 – 400 x 103/ul (x 109/l) poziom hemostatyczny płytek (najmniejsza liczba płytek warunkująca czynność hemostatyczną płytek) PLT = 35-40 x 103/ul poziom zagrażający życiu PLT poniżej 10 x 103/ul małopłytkowość PLT poniżej 100 x 103/ul nadpłytkowość PLT powyżej 600 x 103/ul
FUNKCJE PŁYTEK KRWI
Zespół tenazy Zespół protrombinazy
OSOCZOWE CZYNNIKI KRZEPNIĘCIA KRWI Synteza genetycznie uwarunkowana, geny kodujące syntezę większości czynników zlokalizowane są na chromosomach autosomalnych, z wyjątkiem genów kodujących cz. VIII i IX – w chromosomie płciowym X CZYNNIKI KONTAKTU Białka kofaktorowe , produkowane w wątrobie, niezbędne do aktywacji krzepnięcia w kontakcie z ujemnie naładowanymi powierzchniami: włókna kolagenu, kaolin, szkło Czynnik XII = cz. kontaktu = cz. Hagemana Prekalikreina = Kalikreinogen = cz. Fletschera – proenzym kalikreiny, - aktywator cz. XII i plazminogenu Wielkocząsteczkowy kininogen = cz. Fitzgeralda = HMWK – kofaktor aktywacji cz. XII, XI i kalikreinogenu Czynnik XI = cz. Rosenthala = cz. przeciwhemofilowy- C CZYNNIKI ZESPOŁU PROTROMBINY Synteza w hepatocytach przy udziale witaminy K jako kofaktora. Czynnik X = cz. Stuarta-Prowera Czynnik IX = cz. Christmasa = cz. przeciwhemofilowy -B Czynnik VII = Prokonwertyna = cz. stabilny Czynnik II = Protrombina
CZYNNIKI WRAŻLIWE NA TROMBINĘ synteza w hepatocytach. Czynniki V i VIII są najbardziej labilnymi cz. i szybko ulegają degradacji w próbce krwi przechowywanej w temp. pokojowej lub w podgrzanym osoczu. Czynnik XIII = czynnik stabilizujący fibrynę, transglutaminaza osoczowa Czynnik VIII = czynnik przeciwhemofilowy A, globulina antyhemofilowa Czynnik V = proakceleryna Czynnik I = fibrynogen Dodatkowe czynniki: Czynnik III = tromboplastyna tkankowa = czynnik tkankowy (TF) = białko integralne błon komórkowych m.in. fibroblastów, monocytów, makrofagów, rec. dla cz. VIIa Czynnik IV - jony Ca2+ Cz.vWillebranda – wyst. w osoczu i w płytkach krwi, ułatwia adhezję płytek tworząc kompleks z cz. VIII, chroni go przed proteolityczną degradacją przez (APC) FL- Fosfolipidy błon komórkowych płytek – fosfatydyloseryna
Formacja fibryny Rozpuszczalne Polimery fibryny Protrombina Trombina Fibrynogen monomery fibryny Monomery fibryny fibryna Rozpuszczalne Polimery fibryny
Formacja skrzepu Wiązania krzyżowe Protrombina Trombina Fibrynogen monomery fibryny XIII XIIIa XIII XIIIa fibryna Wiązania krzyżowe
Skrzep Stabilizowana Fibryna powiązana wiązaniami krzyżowymi fibrin
Wiązanie cząsteczek fibryny Wiązania niekowalencyjne Moseson MW, J Lab Clin Med, 1990
Wiązania krzyżowe fibryny IIa XIII XIIIa SKRZEP Wiązania kowalencyjne Moseson MW, J Lab Clin Med, 1990
Moseson MW, J Lab Clin Med, 1990 Fibrynoliza plazmina plazmina plazmina plazmina Moseson MW, J Lab Clin Med, 1990
Aktywacja fibrynolizy tPA fibryna ScuPA F XII, PKK ? DcuPA plazmina plazminogen Terapia fibrynolityczna: SK, t-PA, etc tPA: Tkankowy Aktywator Plazminogenu ScuPA: Single-chain urokinase Plasminogen Activator DcuPA: Double-chain uPA SK: Streptokinaza
Regulacja fibrynolizy tPA fibryna ScuPA F XII, PKK ? PAI-1 DcuPA PAI-1, PAI-2 C1-INH a2-AP, a2-MG plazmina plazminogen HRGP PAI: Inhibitor Aktywatora Plazminogenu C1-inh: C1 inhibitor HRGP: Histidin Rich GlycoProtein 2AP: 2 antyplasmina 2MG: 2 makroglobulina inhibitory
PRODUKTY DEGRADACJI FIBRYNOGENU (FDP) D-DIMER E Fragmenty zawierające D-DIMER Moseson MW, J Lab Clin Med, 1990
PRODUKTY DEGRADACJI FIBRYNOGENU (FDP) FDP-X FDP-D FDP-Y FDP-E FDP-D
PRODUKTY DEGRADACJI FIBRYNOGENU (FDP) DD DY YY XD XY DXD YXD XX YXY XXD Physiologie de la Fibrinolyse, Alessi et al., Manuel d'hémostase, Option Bio, 1996, pp 70 DIC /Martine Migaud-Fressart 1998
Fibrynogen 2 monomeryczne fragmenty D tzw. FDP 1 dimeryczny fragment E plazmina Fibrynogen 2 monomeryczne fragmenty D tzw. FDP 1 dimeryczny fragment E Funkcje FDP hamuje trombinę blokuje czynnik tkankowy hamuje funkcję płytek (adhezję, agregację, r. uwalniania) Norma FDP 0 – 10ug/ml FDP DIC z hiperfibrynolizą Pierwotna hiperfibrynoliza ! (po zabiegach na narządach z t-PA (płuca, macica, stercze) Zakrzepica żył głębokich Białaczki, nowotwory złośliwe Zawał m. sercowego Zator tętnicy płucnej Leczenie np. streptokinazą (wynik oznaczenia FDP obejmuje łącznie produkty degradacji fibrynogenu i fibryny) stabilizowana fibryna 2 fragmenty D połączone wiąz. krzyżowym przez cz. XIII 1 fragment E tzw. D- dimery Funkcje D-dimerów Trawienie zakrzepu D-dimerów DIC , zakrzepica żył głębokich Norma D-dimerów do 200 ng/ml
PRODUKTY ROZPADU FIBRYNY D-DIMERY TO PRODUKTY ROZPADU FIBRYNY
TO MARKERY KRZEPNIĘCIA D-DIMERY TO MARKERY KRZEPNIĘCIA ZACHODZĄCEGO IN VIVO
OBECNOŚĆ D-DIMERÓW W OSOCZU ŚWIADCZY O RÓWNOCZESNEJ AKTYWACJI KRZEPNIĘCIA KRWI I FIBRYNOLIZY
DIC AKTYWACJA WYKRZEPIANIA AKTYWACJA FIBRYNOLIZY
PODWYŻSZONY POZIOM D-DIMERÓW CHOROBA ZAKRZEPOWO-ZATOROWA ZESPÓŁ WYKRZEPIANIA WEWNĄTRZNACZYNIOWEGO NOWOTWORY ZAKAŻENIA CHOROBA WIEŃCOWA I ZAWAŁ SERCA REUMATOIDALNE ZAPALENIE STAWÓW URAZY I OPERACJE POSOCZNICA CIĄŻA OSOBY STARSZE
WPŁYW NA POZIOM D-DIMERÓW MAJĄ: DIAGNOSTYKA SZPITALNA CZY AMBULATORYJNA CZAS TRWANIA HOSPITALIZACJI LECZENIE PRZECIWZAKRZEPOWE (HEPARYNA, DOUSTNE ANTYKOAGULANTY)
WSKAZANIA KLINICZNE DO OZNACZENIA POZIOMU D-DIMERÓW WYKLUCZENIE ZAKRZEPICY ŻYLNEJ LUB TĘTNICZEJ WYKLUCZENIE ZATOROWOŚCI PŁUCNEJ ZESPOŁY WYKRZEPIANIA WEWNĄTRZNACZYNIOWEGO (DIC)
D-DIMER W OSOCZU TO NAJBARDZIEJ PRZYDATNY MARKER LABORATORYJNY DO BADAŃ PRZESIEWOWYCH U OSÓB Z PODEJRZENIEM ZAKRZEPICY ŻYLNEJ LUB ZATOROWOŚCI PŁUCNEJ SZCZEGÓLNIE W WARUNKACH AMBULATORYJNYCH LUB W IZBIE PRZYJĘĆ
DIC - PRZYCZYNY POSOCZNICA URAZY OPERACJE NOWOTWORY UDAR TONIĘCIE HEMOLIZA WEWNĄTRZNACZYNIOWA TĘTNIAK ROZWARSTWIAJĄCY AORTY ODKLEJENIE SIĘ ŁOŻYSKA ZATOR WODAMI PŁODOWYMI OBUMARŁA CIĄŻA CHOROBY TKANKI ŁĄCZNEJ PRZEWLEKŁA NIEWYDOLNOŚĆ NEREK CHOROBA ZAKRZEPOWO-ZATOROWA ZAPALENIE WSIERDZIA
NATURALNE INHIBITORY KRZEPNIĘCIA utrzymanie płynności krwi krążącej oraz zapobieganie nadmiernemu narastaniu czopu ostatecznego najważniejsze inhibitory: Antytrombina III – AT III Białko C (PC) Białko S (PS) jako kofaktor białka C Inhibitor zewnątrzpochodnego szlaku krzepnięcia –TFPI ANTYTROMBINA III - synteza w wątrobie, w mniejszym stopniu w śródbłonku naczyń oraz w megakariocytach - należy do rodziny serpin inaktywujących proteazy serynowe poprzez ich wiązanie w kompleks stechiometryczny w stos. 1:1 - kofaktorem AT III jest heparyna, która tworząc z nią kompleks przyśpiesza reakcję inaktywacji cz. krzepnięcia 1000 x. Trombinę cz. XIIa AT III inaktywuje : cz. XI a cz. IX a cz. Xa, (ostatnio uważa się, że również VIIa)
BIAŁKO C - synteza formy nieaktywnej w wątrobie przy udziale witaminy K - aktywacja białka C uruchamiana jest przez pojawienie się trombiny we krwi trombina wiąże się w kompleks z trombomodulina inaktywacja cz. VIII a Białko C APC + PS inaktywacja cz. V a Niedobór białka C : DIC leczenie L-asparaginazą, doustnymi antykoagulantami TFPI - inhibitor zewnątrzpochodnego szlaku krzepnięcia - białko występujące w osoczu w formie związanej z Lp - w obecności cz. Xa wiąże i inaktywuje kompleks TF – VII a
ZASADY POBIERANIA KRWI DO BADAŃ UKŁADU KRZEPNIĘCIA RANO, NA CZCZO ( lub lekki posiłek beztłuszczowy) W WARUNKACH SPOKOJU, BEZ STRESU KREW ŻYLNA NA 3,2% CYTRYNIAN SODU (1 cz. cytrynianu - 9 cz. krwi ) BEZ STAZY lub UCISK ok. 30 sek OSTRE, DOŚĆ GRUBE IGŁY KREW PO ODRZUCENIU PIERWSZYCH 2-3 ML PLASTIKOWE PROBÓWKI BARDZO DELIKATNE MIESZANIE KRWI TUŻ PO POBRANIU (3-4 krotne odwrócenie probówki do góry dnem - nie wolno spienić! ) NIEZWŁOCZNIE ODWIROWAĆ PRÓBKĘ KRWI (10 min – 3 tys. obrotów/min) BADANIA WYKONAĆ W CIĄGU MAX 2 GODZIN OD POBRANIA
Czas krwawienia metodą Ivy BADANIA PRZESIEWOWE (PODSTAWOWE) I UZUPEŁNIAJĄCE W DIAGNOSTYCE ZABURZEŃ UKŁADU HEMOSTAZY NACZYNIOWE Czas krwawienia metodą Ivy Testy specjalistyczne – test oporności kapilarowej - biopsja skóry PŁYTKOWE Czas krwawienia - BT Liczba płytek krwi –PLT Rozmaz krwi obwodowej Badania czynnościowe: Adhezja i agregacja płytek Reakcja uwalniania Ocena zawartości ziarnistości płytek Czas zużycia protrombiny OSOCZOWE Czas kaolinowo-kefalinowy – aPTT – (k-k) -czas częściowej tromboplastyny po aktywacji Czas protrombinowy – PT Czas trombinowy – TT Czas rekalcynacji – CT Fibrynogen Poszczególne czynniki krzepnięcia: VIII, VII, XIII Krążące antykoagulanty Inhibitory krzepnięcia: Antytrombina III –AT III Białko C, białko S
Czas od chwili uszkodzenia naczynia do samoistnego ustania krwawienia CZAS KRWAWIENIA Czas od chwili uszkodzenia naczynia do samoistnego ustania krwawienia jest jednoznaczny z czasem trwania hemostazy pierwotnej jest miarą: czasu utworzenia czopu płytkowego skurczu naczyń adhezji i agregacji płytek do śródbłonka naczyń próba czynnościowa płytek krwi zależna częściowo od stanu naczyń, nie zależy od czynników krzepnięcia metody oznaczania : Metoda Duke’a Nakłucie skóry opuszki palca na głębokość 2-3 mm i pomiar czasu do momentu zaprzestania wypływu krwi Norma - do 5 min
Metoda Ivy z modyfikacją Mielke – met. zalecana Standaryzacja pomiaru – pomiar czasu wypływu krwi od momentu dokonania nacięcia na skórze przedramienia o dł. 10 mm i głębokości ok. 2,5 mm (zestawy np. Simplate) przy ciśn. 40 mmHg (ciśn. nadmuchane po założeniu opaski ciśnieniomierza) do braku śladu krwi na przykładanej do nacięcia bibule filtracyjnej. Norma – 2 – 10 min (do 8 min) CZAS KRWAWIENIA małopłytkowość trombastenia Glanzmana (wrodzone zaburzenia agregacji płytek) ch. von Willebranda niektóre trombocytopatie skazy krwotoczne z hipofibrynogenemią po aspirynie
CZAS REKALCYNACJI OSOCZA - CK czas krzepnięcia po dodaniu do osocza nadmiaru jonów Ca2+ w probówce szklanej ocenia aktywność układu wewnątrzpochodnego Norma CT: 100 – 210 sek próba mało czuła, czas wydłuża się dopiero po zmniejszeniu aktywności jakiegoś czynnika poniżej 3 – 5% normy ( skazy krwotoczne wyst. już przy wartościach 10 – 20 % normy) CZAS KRZEPNIĘCIA WG. METODY LEE – WHITE’A (CZAS L-W) pomiar czasu od pobrania pełnej krwi do jej skrzepnięcia w szklanej probówce w temp. 37 C określa sprawność układu wewnątrzpochodnego (próba mało czuła, czas wydłuża się dopiero po zmniejszeniu aktywności jakiegoś czynnika poniżej 1 – 3 % normy) Norma czasu L-W : 8 – 12 min Czas krzepnięcia · niedobór cz. układu wewnątrzpochodnego (XII, XI, IX, VIII) · niedobór cz. drogi wspólnej ( X, V, II, I) · obecność inhibitorów czynników krzepnięcia np. podczas leczenia heparyną
PT – ocena zewnątrzpochodnego układu krzepnięcia Czynnik VII Czynnik VII a, cz. tkankowy (TF) Ca 2+ Czynnik X czynnik X a czynnik Va, Ca 2+, FL protrombina trombina fibrynogen fibryna SPOSOBY WYRAŻANIA CZASU PROTROMBINOWEGO tromboplastyna 100 ul osocza 100 ul tromboplastyny 100 ul CaCl2 CZAS PROTROMBINOWY Czas (sekundy) 11 – 13 sek 12 – 16 sek WSKAŹNIK PROTROMBINOWY PT prawidłowy x 100 PT pacjenta 80 – 120 % WSPÓŁCZYNNIK PROTROMBINOWY - R PT prawidłowy 0,85 – 1,15 INR – międzynarodowy współczynnik znormalizowany R ISI ISI – międzynarodowy indeks czułości 0,9 – 1,25
TT – ocena przejścia fibrynogenu w fibrynę – ocena drogi wspólnej Norma TT – ok. 15 sek Trombina TT zależy od: stęż. Fbg, trombiny, aktywności AT III, prawidłowej stabilizacji fibryny
TT · Hipofibrynogenemia np. w marskości wątroby, DIC i afibrynogenemii 0 g/l · Dysfibrynogenemia · Obecność inhibitorów trombiny np. leczenie heparyną · Obecność inhibitorów polimeryzacji fibryny np. FDP · Inne: mocznica, gammapatia monoklonalna TT · Nadkrzepliwość TT wykonać Czas reptylazowy pomiar czasu krzepnięcia osocza po aktywacji reptylazą (wyciąg z jadu węża) - enzym o działaniu podobnym do trombiny, ale niezależny od heparyny norma – ok. 18 –20 sek TT + t. reptylazowy obecność FDP (hamują polimeryzację fibryny) TT + N t. reptylazowy obecność heparyny (hamuje trombinę (nie hamuje reptylazy)
FIBRYNOGEN - FBG Glikoproteina syntetyzowana w wątrobie niezbędna w procesie krzepnięcia do tworzenia czopu płytkowego (warunkuje agregację płytek) i ostatecznego (tworzenie siatki fibrynowej) Białko ostrej fazy, którego stęż. rośnie w początkowym okresie infekcji (wpływ na wzrost OB) niezależny czynnik ryzyka choroby wieńcowej, zawału m. sercowego, udaru mózgu (bierze udział w transporcie chol i tworzeniu k. piankowatych, powoduje rozrost mięśni gładkich – zw. miażdżycorodny) wzrost między 3-5 dniem w zawale, stabilizacja w ciągu 20 dni im wyższy fbg, tym gorsze rokowanie w zawale Norma - 2,0 – 5,0 g/l 200-500 mg/dl Metody oznaczania fbg: metoda chronometryczna Claussa (ścisła korelacja między trombinowym czasem krzepnięcia i stężeniem fibrynogenu w osoczu) metoda kolorymetryczna z odczynnikiem Folina i Ciocalteu
Fibrynogen DIC pierwotna hiperfibrynoliza wrodzony brak lub niedobór fbg marskość wątroby i inne uszkodzenia czynności wątroby ch. hematologiczne (AL, nk aplastyczna) podczas leczenia streptokinazą hiperfibrynogenemia przemijająca: Hiperfibrynogenemia dłużej trwająca III trymestr ciąży i okres okołoporodowy nowotwory złośliwe po zabiegach operacyjnych przewlekłe stany zapalne zakrzepica kolagenozy urazy i ostre stany zapalne, zwłaszcza zapalenie płuc zespół nerczycowy zawał serc
FIBRYNOLIZA SKRZEPU EUGLOBULIN OSOCZA - próba ogólna układu fibrynolitycznego - czas upłynnienia skrzepu euglobulin osocza wytrąconych pod wpływem roztworu o niskiej sile jonowej, pH-5 i temp. 4C, a następnie rozpuszczonych w buforze i wykrzepionych CaCl2 - mierzy się czas od momentu powstania skrzepu do chwili rozpuszczenia w temp. 37C - euglobulinowa frakcja białkowa zawiera : plazminogen plazmina aktywatory plazminogenu fibrynogen cz. krzepnięcia: m.in. VIII, XII, XIII - brak inhibitorów plazminogenu np. antyplazmin (które pozostają w supernatancie) Norma - 120 – 240 min (2 – 4 godz. )
czas lizy euglobulin III trymestr ciąży okres pooperacyjny - choroby zatorowo-zakrzepowe - zawał serca i miażdżyca - cukrzyca - nadciśnienie skazy krwotoczne przebiegające z hiperfibrynolizą - zaawansowana choroba nowotworowa - marskość wątroby - ostre stany septyczne - po przetoczeniu krwi obcej grupowo przy znacznym skróceniu czasu lizy euglobulin przed operacją podaje się leki hamujące aktywność t-PA
ALGORYTM CZASÓW
obecność krążącego antykoagulantu próba korekcji APTT APTT PT - N, TT - N , BT - N niedobór cz. XII, XI, IX, VIII (hemofilia lub ch. vW), PK obecność krążącego antykoagulantu próba korekcji APTT prawidłowa brak korekcji niedobór czynników krążący antykoagulant (anty-VIII lub LA) oznaczyć poziom poj. czynników miano APTT PT TT – N, BT -N niedobór cz. II, V, X, VII (choroby wątroby) złożony niedobór cz. zależnych od witaminy K (znaczny) zaburzenia po masywnych przetoczeniach próba korekcji APTT oznaczyć poziom cz. II, V, VII, X APTT PT TT BT – N hypo- i dysfibrynogenemie DIC ch. wątroby oznaczyć stęż. fibrynogenu aktywacja fibrynolizy FDP wpływ heparyny czas reptylazowy APTT – N PT TT – N, BT – N niedobór cz. VII rozpoczęcie leczenia antykoagulantami doustnymi APTT- N, PT – N, TT- N, BT ch. von Willebranda APTT – N, PT – N, TT- N, BT zaburzenia płytkowo-naczyniowe
PODZIAŁ SKAZ KRWOTOCZNYCH NACZYNIOWE Wrodzone: · wrodzona naczyniakowatość krwotoczna (ch. Rendu-Oslera) · plamice we wrodzonych zaburzeniach tkanki łącznej np. zespół Marfana · samoistna hemosyderoza płuc Nabyte: · zespół Schönleina-Henocha · plamice w przebiegu zakażeń · plamice w przebiegu amyloidozy · plamica starcza, ortostatyczna, mechaniczna, polekowe, z zapaleniem naczyń włosowatych, szkorbut - rozpoznanie : przedmiotowe bad. lekarskie + wywiad rodzinny wyniki testów z zakresu krzepnięcia krwi i hemostazy - norma
PŁYTKOWE Zmiany ilościowe: A. Małopłytkowości - Trombocytopenie a) zmniejszone wytwarzanie płytek wrodzone: * wrodzona hipoplazja szpiku – zespół Fanconiego nabyte : * nk aplastyczna * aplazja megakariocytowa * nacieczenie szpiku kostnego (ALL, chłoniaki, gruźlica) * zwłóknienie szpiku * leki mielosupresyjne, zw. chemiczne, promienie jonizujące * nk megaloblastyczne, z niedoboru żelaza, nocna napadowa hemoglobinuria * zakażenia wirusowe, niewydolność nerek b) nadmierne niszczenie płytek wrodzone: * samoistna autoimmunologiczna plamica małopłytkowa nabyte: immunologiczne: * małopłytkowość poprzetoczeniowa, polekowa * autoimmunologiczna nk hemolityczna * toczeń rumieniowaty układowy * wstrząs anafilaktyczny
B. Nadpłytkowości nieimmunologiczne: * DIC * zakażenia * zespół hemolityczno- mocznicowy * zakrzepowa plamica małopłytkowa c) nieprawidłowe rozmieszczenie płytek w ustroju * hipersplenizm d) utrata płytek: * krążenie pozaustrojowe, krwotoki B. Nadpłytkowości pierwotne: * samoistna, * zespóły mieloproliferacyjne wtórne: * stany zapalne (gruźlica, sarkoidoza, RKZ, wrzodziejące zap. jelita grubego) * ch. nowotworowe * po splenektomii i innych zabiegach pooperacyjnych * po krwotokach * w niedoborze żelaza * polekowa ( np. winkrystyna) * powysiłkowa (trwająca ok. 15-30 min)
Zmiany jakościowe (zaburzona czynność płytek)- TROMBOCYTOPATIE: Wrodzone a) a) anomalie błony płytkowej: * zespół Bernarda-Souliera (defekt kompleksu GP Ib/IX/V- płytkowy rec. dla cz.vW zaburzona adhezja płytek krwi * trombastenia Glanzmanna ( defekt kompleksu GP IIb/IIIa – rec. dla fbg) zaburzona agregacja płytek krwi * defekt receptora kolagenu ( defekt GP Ia/IIa) - łagodna skaza krwotoczna * zespół Scotta (zaburzenia prokoagulacyjnej czynności płytek) b) zaburzenia sekrecji ziarnistości płytkowych B. Nabyte : * wpływ leków * ch. krwi (ALL, MDS, zespoły mieloproliferacyjne), * DIC * inne choroby np. mocznica, krążenie pozaustrojowe, przewlekłe ch. wątroby
OSOCZOWE Wrodzone: * Ch. von Willebranda * Hemofilia A *Hemofilia B * A-, hipo-, dysfibrynogenemia * Niedobór pojedynczego cz. II, V, VII, X, XI, XII, XIII (występują rzadko) Nabyte: a) a) niedobór witaminy K upośledzenie wytwarzania wit. K np. * ch. krwotoczna noworodków upośledzenie wchłaniania wit.K *kamica, nowotwór, zespół złego wchłaniania upośledzone wykorzystanie wit. K * doustne antykoagulanty-pochodne dihydroksykumaryny b) zaburzenia krzepnięcia z powodu chorób wątroby
FIBRYNOLITYCZNE Czas lizy euglobulin Czas trombinowy FDP, D-dimery Plazminogen Aktywatory fibrynolizy: Tkankowy aktywator plazminogenu – t-PA Urokinaza – u-PA Inhibitory fibrynolizy: Inhibitor aktywatora plazminogenu – PAI-1 2 antyplazmina - 2 AP
TROMBOFILIA – NADKRZEPLIWOŚĆ Wrodzona lub nabyta skłonność do zakrzepów przyczyny zaburzeń nabytych: obecność p/c antyfosfolipidowych (Lupus antykoagulant- LA) nadpłytkowość czerwienica prawdziwa zwiększona aktywność inhibitorów fibrynolizy przyczyny wrodzonej trombofilii ( wyst. poniżej 40 r.ż., bez czynników ryzyka): oporność na aktywne białko C (APC-R) – 12 –64 % częstość występowania niedobór białka C - 5 –8 % niedobór białka S - 5 – 8 % niedobór AT III - 2 – 4 % APC-R zaburzenie związane z mutacją punktową w obrębie cz. V typu Leiden, zmieniony cz. V ma aktywność prokoagulacyjną, natomiast nie ulega proteolizie pod wpływem APC
HHEMOFILIE - skaza krwotoczna osoczowa wwrodzone zaburzenie krzepnięcia krwi związane z chromosomem X (dziedziczenie recesywne sprzężone z płcią - chorują chłopcy, kobiety są nosicielami, chorują przy spadku poziomu czynnika VIII poniżej 40 %): hemofilia A – wrodzony niedobór cz. VIII hemofilia B – wrodzony niedobór cz. IX hemofilia C – wrodzony niedobór cz. XI HEMOFILIA A - wrodzony niedobór cz. VIII, stopień ciężkości choroby zależy od aktywności cz. VIII Postać hemofilii Aktywność cz. VIII w surowicy Objawy ciężka poniżej 1 % częste nawracające krwawienia do stawów (zniekształcenia), do mięśni i narządów wewnętrznych średnio ciężka 1 – 5 % krwawienia samoistne (rzadko) Ciężkie krwawienia po ekstrakcji zębów, zabiegach chirurgicznych, krwiaki pourazowe łagodna 5 – 15 % Krwawienia po zabiegach, urazach subhemofilia 15 – 30 % bezobjawowo
Rozpoznanie hemofilii - objawy kliniczne + wywiad rodzinny - badania : APTT - ! - sprawdzić aktywność cz. VIII PLT - N prawidłowa hemostaza pierwotna (brak wybroczyn na skórze) Czas krwawienia – N PT - N Podanie dawki cz. VIII Dawka = m.ciała (kg) x pożądany wzrost aktywności (w %) x 0,5 Podanie preparatu – sprawdzenie APTT brak skrócenia Obecność autoprzeciwciał przeciw cz. VIII lub krążącego antykoagulanta
DIAGNOSTYKA LABORATORYJNA DIC FDP Fibrynogen i FDP – wysoce czułe w diagnostyce DIC Cut-off ustalony dla zespołu DIC D-Dimer Uzupełnienie testu oceniającego poziom FDP Bardzo czuły (norma < 0.5 µg/ml) Łączna ocena wyników obu testów daje bardzo wysoką trafność diagnostyczną w rozpoznawaniu zespołu DIC
DIAGNOSTYKA LABORATORYJNA DIC INNE TESTY LABORATORYJNE ATIII TAT PAP KOFAKTOR HEPARYNY II CZYNNIKI VIII, V, VII PF3, PF4 PLAZMINOGEN BIAŁKO C PŁYTKI SCHISTOCYTY
pobieranie krwi: bezpośrednio przed podaniem heparyny po 4-5 godz – w przypadku wlewu ciągłego po 4 godz - przy podaniu podskórnym terapeutyczne stęż. heparyny – 1,5 - 2,5 x wzrost APTT w porównaniu z APTT przed leczeniem – 1,5 – 3 x wzrost CT w porównaniu z CT przed leczeniem Objawy uboczne UFH : małopłytkowość na poczatku leczenia, utajony wrzód żołądka lub dwunastnicy, skaza krwotoczna, osteoporoza (po okresie 3 miesięcy) (hirudyna -wyciąg ze ślinianek pijawek lekarskich -działa bezpośrednio na AT III ANTYKOAGULANTY DOUSTNE - hamują cykl przemian wit K hamowanie produkcji cz. wit. K zależnych (II, VII, IX, X) hamują produkcję białka C i S monitorowanie leczenia poprzez pomiar PT terapeutyczne stęż. leku pobieranie krwi: bezpośrednio przed podaniem INR – 2,0 – 3,0 w 3, 5, 7 dniu leczenia, INR – 2,5-3,5 (w ciężkich przypadkach) co tydzień w pierwszym m-cu leczenia co 2-3 tyg. w II i III m-c co 4-6 tyg w okresie póżniejszym