Pobierz prezentację
1
Współczesne metody analiz genetycznych
Anna Jakubowska Katedra Onkologii Zakład Genetyki i Patomorfologii PUM

2
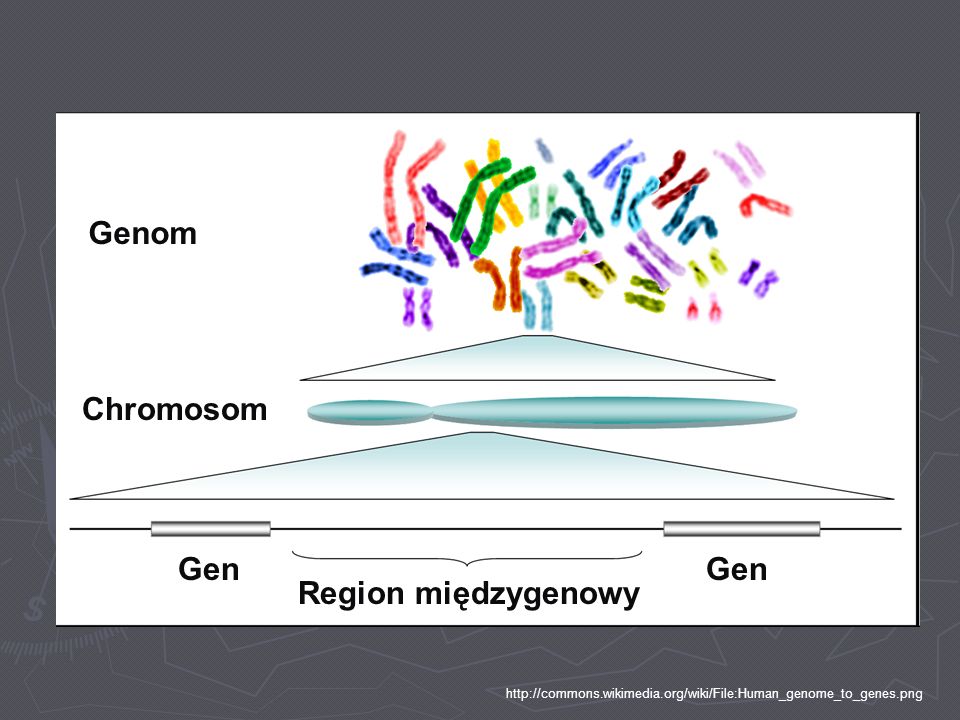
Genom Chromosom Gen Gen Region międzygenowy
3
Poznanie genomu człowieka
rozpoczęcie HGP – (Human Genome Project) wstępny opis sekwencji – 2000 (Venter et al., Science 2001; Lander et al., Nature 2001) zakończenie sekwencjonowania – 2003 oficjalne zakończenie HGP – (International Human Genome Sequencing Consortium, Nature 2004 )
 wstępny opis sekwencji – (Venter et al., Science 2001; Lander et al., Nature 2001) zakończenie sekwencjonowania – oficjalne zakończenie HGP – 2004 (International Human Genome Sequencing Consortium, Nature 2004 )")
4
Struktura genomu człowieka
pz ~ 2% to sekwencje kodujące genów kodujących białka 8 483 genów kodujących RNA pseudogenów ~ 98% to sekwencje niekodujące introny oraz sekwencje międzygenowe SNPs (polimorfizmy pojedynczego nukleotydu) ~ regionów CNV (duplikacje lub delecje odcinków DNA o długości >500 zasad) (
 ~ regionów CNV (duplikacje lub delecje odcinków DNA o długości >500 zasad) (")
5
Geny funkcja wielu genów wciąż niepoznana
identyfikacja genów lub specyficznych stref genetycznych związanych z rozwojem chorób: badania asocjacyjne sekwencjonowanie genomu

6
Badania asocjacyjne analiza wybranych markerów –kandydatów
analiza markerów pokrywających cały genom, tzw. GWAS (Genome – Wide Association Study) potocznie nazywana skanowaniem genomu
 potocznie nazywana skanowaniem genomu.")
7
GWAS Badanie „przypadek - kontrola” („case - control”), w którym analizuje się związek pomiędzy występowaniem określonej cechy klinicznej a SNPs lub CNVs rozmieszczonymi w całym genomie A B C A B B B C A C SNP CNV
, w którym analizuje się związek pomiędzy występowaniem określonej cechy klinicznej a SNPs lub CNVs rozmieszczonymi w całym genomie. A. B. C. A. B. B. B. C. A. C. SNP. CNV.")
8
Założenia GWAS badanie dużej liczby polimorfizmów o największej zmienności międzyosobniczej badanie jak największej grupy osób chorych i zdrowych

9
Identyfikacja SNP związanych z chorobą
Chorzy Zdrowi DNA DNA Porównanie Identyfikacja SNP związanych z chorobą

10
Badania GWAS – kluczowe zasady
homogenność grupy badanej pod względem analizowanej cechy liczna grupa kontrolna oraz badana walidacja

11
Etapy GWAS Przypadki i kontrole Badane SNPs I etap II etap III etap
Multistage design for genome-wide association studies. In a multistage design, a large number of SNPs selected to capture most common genetic variations across the genome (genome-wide scan chip) are tested in a relatively small number of cases and controls in a “discovery study.” The SNPs showing the most significant associations with disease risk in the discovery study (e.g., P value from an association test <0.05) are retested in subsequent replication studies including large independent sets of cases and controls. In the example shown in the figure, SNPs with P values <0.001 in a first replication study are retested in a second replication study. SNPs showing strong evidence of an association with disease risk based on data from the three phases (e.g., P value from an association test <10−7) are selected as markers for chromosomal regions likely to contain disease causing variants. Very large studies and stringent statistical criteria are necessary to have sufficient power to detect associations while minimizing the probability of false-positive findings. The selected markers in genome-wide association studies are further evaluated in fine mapping studies to identify causal variants, and in functional studies to understand the biological mechanism of the observed associations with disease. Red and blue individuals represent cases of breast cancer and controls, respectively, being tested at different stages of the design. Green and red dots in the inverted cone represent SNPs being tested at each stage. The red dots are markers for disease susceptibility alleles. Mapowanie zmian Analiza funkcjonalna Garcia-Closas M , Chanock S Clin Cancer Res 2008;14: ©2008 by American Association for Cancer Research
 are tested in a relatively small number of cases and controls in a discovery study. The SNPs showing the most significant associations with disease risk in the discovery study (e.g., P value from an association test <0.05) are retested in subsequent replication studies including large independent sets of cases and controls. In the example shown in the figure, SNPs with P values <0.001 in a first replication study are retested in a second replication study. SNPs showing strong evidence of an association with disease risk based on data from the three phases (e.g., P value from an association test <10−7) are selected as markers for chromosomal regions likely to contain disease causing variants. Very large studies and stringent statistical criteria are necessary to have sufficient power to detect associations while minimizing the probability of false-positive findings. The selected markers in genome-wide association studies are further evaluated in fine mapping studies to identify causal variants, and in functional studies to understand the biological mechanism of the observed associations with disease. Red and blue individuals represent cases of breast cancer and controls, respectively, being tested at different stages of the design. Green and red dots in the inverted cone represent SNPs being tested at each stage. The red dots are markers for disease susceptibility alleles. Mapowanie zmian. Analiza funkcjonalna. Garcia-Closas M , Chanock S Clin Cancer Res 2008;14: ©2008 by American Association for Cancer Research.")
12
Polimorfizmy dla GWAS wysoka częstość, MAF (minor allele frequency) >5% ~7 mln SNP o częstości MAF >5% ~4 mln SNP o częstości 1-5% brak sprzężenia SNP nie powinny znajdować się w genomie blisko siebie w tzw. nierównowadze sprzężeń (linkage disequilibrium) wykorzystanie mikromacierzy Affymetrix Illumina
 wykorzystanie mikromacierzy. Affymetrix. Illumina.")
13
Projekt HapMap międzynarodowe przedsięwzięcie obejmujące Japonię, Kanadę, Chiny, Nigerię i USA w celu identyfikacji oraz określenia częstości zmian polimorficznych w genomie ludzkim w różnych populacjach 270 osób: 90 z Nigerii 45 z Japonii 45 z Chin 90 z Europy północnej i zachodniej

14
Analiza mikromacierzy
wytworzenie mikromacierzy naniesienie na płytkę gotowych lub zsyntetyzowanych in situ sond długości nukleotydów mikromacierz Sonda oligonukleotydowa

15
Analiza mikromacierzy
hybrydyzacja DNA na płytce zawierającej sondy

16
Analiza mikromacierzy
skanowanie

17
Analiza mikromacierzy
analiza ilościowa

18
Analiza mikromacierzy
Affymetrix Illumina

19
Affymetrix „chip” strategia wyboru SNP: sondy detekcja
Affymetrix's Genome-Wide Human SNP Array 6.0 zawiera sondy dla SNP i CNV strategia wyboru SNP: tylko połowa w oparciu o projekt HapMap, tzw. haplotype - tagging „tagSNPs”, reszta to dowolne SNPs występujące w genomie sondy 25-nukleotydowe, 4-6 kopii/zmianę detekcja hybrydyzacja wyznakowanego DNA

20
Trawienie enzymami restrykcyjnymi
Ligacja z adapterem Nsp lub Sty Amplifikacja PCR z jednym starterem Oczyszczenie próbki Fragmentacja, znakowanie końców Hybrydyzacja

21
Illumina „chip” HumanOmni2.5-Quad BeadChip zawiera > 2.45 milliona sond do badania SNPs i CNVs strategia wyboru SNPs: w oparciu o projekt HapMap, tzw. haplotype - tagging „tagSNPs” sondy 50-nukleotydowe, 1/zmianę detekcja wydłużanie sond z dobudowaniem znakowanych nukleotydów

22
Hybrydyzacja DNA z sondą
Genomowe DNA ( ng) Amplifikacja DNA Fragmentacja DNA 2-etapowa detekcja Etap I Hybrydyzacja DNA z sondą Etap II Wydłużanie sond z dobudowaniem wyznakowanych nukleotydów
 Amplifikacja DNA. Fragmentacja DNA. 2-etapowa detekcja. Etap I. Hybrydyzacja DNA z sondą. Etap II. Wydłużanie sond z dobudowaniem wyznakowanych nukleotydów.")
23
Zestawienie wykonanych GWAS
~ 600 GWAS obejmujących różne grupy i populacje 150 różnych zespołów np. cukrzyca typu I i II, choroba Crohna, choroba Alzheimera, schizofrenia, udar, choroby serca, nowotwory złośliwe, otyłość > SNP wyselekcjonowanych do walidacji zidentyfikowano ~800 SNP związanych z chorobami (p<5×10-8) Johnson and O'Donnell, BMC Medical Genetics 2009 Manolio, NEJM 2010
 Johnson and O Donnell, BMC Medical Genetics Manolio, NEJM")
24
Manolio et al., NEJM 2010

25
Badanie Wellcome Trust 2005-2007
8 ważnych schorzeń wieloczynnikowych osób (chorych i zdrowych) polimorfizmów (SNPs i CNVs) 200 badaczy, 9 milionów funtów Nature 2007, Nature 2010
 polimorfizmów (SNPs i CNVs) 200 badaczy, 9 milionów funtów. Nature 2007, Nature")
26
Wyniki badań Wellcome Trust
Choroba afektywna dwubiegunowa locus 16p12 (OR 2,08) Choroba wieńcowa locus 9p21 (ORhet-hom 1,47-1,9) Choroba Crohna 5 znanych lokalizacji (ORhet-hom): NOD2 (1,29-1,92), IL23R (1,39-1,86), ATG16L1 (1,19-1,85), ZNF365 (1,23-1,55), locus 5p13.1 (1,54-2,32) 4 nowe lokalizacje (ORhet-hom): IRGM (1,54-1,92), BSN (1,09-1,84), NKX2-3 (1,2-1,62), PTPN2 (1,3-2,01)
 Choroba wieńcowa. locus 9p21 (ORhet-hom 1,47-1,9) Choroba Crohna. 5 znanych lokalizacji (ORhet-hom): NOD2 (1,29-1,92), IL23R (1,39-1,86), ATG16L1 (1,19-1,85), ZNF365 (1,23-1,55), locus 5p13.1 (1,54-2,32) 4 nowe lokalizacje (ORhet-hom): IRGM (1,54-1,92), BSN (1,09-1,84), NKX2-3 (1,2-1,62), PTPN2 (1,3-2,01)")
27
Wyniki badań Wellcome Trust
Nadciśnienie brak wyraźnych czynników ryzyka - kilka wariantów o stosunkowo małym wpływie (OR 0,97-1,6) Reumatoidalne zapalenie stawów 9 znanych już lokalizacji (OR 0,91-2,3) PTPN22 (ORhet-hom 1,98-3,32), region MHC (ORhet-hom 2,36-5,21) korelacja z chorobami serca i cukrzycą typu I
 Reumatoidalne zapalenie stawów. 9 znanych już lokalizacji (OR 0,91-2,3) PTPN22 (ORhet-hom 1,98-3,32), region MHC (ORhet-hom 2,36-5,21) korelacja z chorobami serca i cukrzycą typu I.")
28
Wyniki badań Wellcome Trust
Cukrzyca typu I 5 znanych lokalizacji, w tym gen PTPN22 (ORhet-hom 1,82-5,19) i MHC (ORhet-hom 5,49-18,52) 3 nowe loci (ORhet-hom ): 12q13 (1,34-1,75), 12q24 (1,34-1,94), 16p13 (1,19-1,55) Cukrzyca typu II TCFL2 (ORhet-hom 1,36-1,88), FTO (ORhet-hom 1,34-1,55), CDKAL1/CDKARAP1 (ORhet-hom 1,18-2,17)
 i MHC (ORhet-hom 5,49-18,52) 3 nowe loci (ORhet-hom ): 12q13 (1,34-1,75), 12q24 (1,34-1,94), 16p13 (1,19-1,55) Cukrzyca typu II. TCFL2 (ORhet-hom 1,36-1,88), FTO (ORhet-hom 1,34-1,55), CDKAL1/CDKARAP1 (ORhet-hom 1,18-2,17)")
29
Znaczenie medyczne identyfikacja grup zwiększonego ryzyka zachorowania na określone choroby związek z pojedynczym markerem, tzw. „single effect” sumowanie ryzyka, tzw. „additive effect” np. OR 1,62 (1 SNP) i OR 9,46 (≥5 SNPs) dla raka prostaty Pharoah et al. NEJM 2008 Zheng et al. NEJM 2008
 i OR 9,46 (≥5 SNPs) dla raka prostaty. Pharoah et al. NEJM Zheng et al. NEJM")
30
Zmiany zidentyfikowane w GWAS
funkcjonalne niefunkcjonalne markery genetyczne identyfikują obszar/gen gdzie należy szukać właściwych mutacji

31
Sekwencjonowanie DNA Technika umożliwiająca ustalenie kolejności zasad w DNA.

32
Sangera – 1975 rok Maxama-Gilberta – 1977 rok
polega na terminacji łańcucha DNA przy wykorzystaniu zmodyfikowanych nukleotydów (dideoksynukleotydy) Maxama-Gilberta – 1977 rok polega na wykorzystaniu związków chemicznych do rozszczepiania łańcucha DNA
 Maxama-Gilberta – 1977 rok. polega na wykorzystaniu związków chemicznych do rozszczepiania łańcucha DNA.")
33
oparte na metodyce Sangera
automatyczne – 1987 rok oparte na metodyce Sangera

34
3730xl DNA Analyzer 96 kapilar 3 840 próbek/dobę 2 100 000 zasad/dobę
do 900 zasad/próbkę

35
pirosekwencjonowanie – 1996 rok
„sekwencjonowanie poprzez syntezę” w reakcji wykorzystywane są cztery enzymy: fragment Klenowa polimerazy DNA I sulfurylaza ATP lucyferaza apiraza.

36
PyroMark Q96 MD do 96 próbek analizowanych jednocześnie
max zasad/próbkę do 960 próbek/dobę krótka procedura przygotowania próbek

37
Sekwencjonowanie nowej generacji
Analiza setek milionów lub miliardów par zasad w jednym ciągu reakcji, przy jednoczesnym obniżeniu kosztów System 454 (Roche) HiSeq (Illumina) SOLiD (Applied Biosystems)
 HiSeq (Illumina) SOLiD (Applied Biosystems)")
38
System 454 w oparciu o pirosekwencjonowanie >1mln zasad/analizę
1 mld zasad/dobę do 450 zasad/próbkę czułość 99% koszt $/analizę

39
HiSeq2000 w oparciu o odwracalną reakcję terminacji
jednoczesna analiza dwóch różnych próbek 200 mld zasad/analizę, 8 dni do 25 mld zasad/dobę do 100 zasad/próbkę czułość >99% koszt $/analizę

40
5500xl SOLiD™ System w oparciu o ligację komplementarnych frag.
jednoczesna analiza 12 różnych próbek 300 mld zasad/analizę, 7 dni do 45 mld zasad/dobę do 75 zasad/próbkę czułość 99.99% koszt $/analizę

41
Postęp w sekwencjonowaniu

42
Podsumowanie najlepsze efekty daje połączenie kilku metod
konieczne nowej jakości badania strukturalne i asocjacyjne : RNA białek innych oddziaływań ze środowiskiem o nieznanym dotąd charakterze






















>")