Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Odlewnictwo Krystalizacja i krzepnięcie
Instytut Materiałów Inżynierskich i Biomedycznych Zakład Odlewnictwa Wydział Mechaniczny Technologiczny POLITECHNIKA ŚLĄSKA W GLIWICACH Odlewnictwo Krystalizacja i krzepnięcie GLIWICE – ROK 2005 Mirosław Cholewa

2
Literatura: Crosley P., Douglas A., Mondolfo L.: Interfacial Energies in Heterogeneous Nucleation,The Solid. of Metals, The Iron and Steel Inst. nr 110, London 1968, s.10 Bramfitt B.: The effect of Carbide and Nitride Addition on the heterogeneous Nucleation Behavior of Liquid Iron, Metall. Trans., 1970, s. 1987 Gliksman M., Childs W.: Nucleation Catalysis in Supercooled Liquid Tin, Acta Metall. 1962, s.925 Fraś E.: Teoret. Podst. Kryst., cz. 1, skr. uczeln. nr 930 Kraków 1984, s 82Oostdijk J., C.: Van kiem tot kristalliet. Pr. Dokt. Delft, Drukkerij J., H. Pasmans, 1967 Chalmers B.: Principles of Solidification, John Wiley & Sons, New York 1964, s.68 Verma A. R.: Crystal growth and dislocations. Butterworths Scientific Publications, London 1953, Missol W.: Energia powierzchni rozdziału faz, Wyd. „Śląsk”. Katowice 1975 Braszczyński J., Krystalizacja odlewów, WNT, Warszawa 1991 Jones H., Leak M.G.: The surface entropy of solid metals, Metall. Sci. v. 1, 1967, s. 211 Jones H.: The surface energy of solid metals. Metall. Sci. v. 5, 1971, s. 15 Jones H.: The grain boundary term in surface energy measurement by zero creep, Scripta Met. v. 6, 1972, s. 423Allen B.C.: Effect of crystallographic orientation on the surface free energy and surface self-diffusion of solid molybdenum. Trans. Metall. Soc. AIME v. 245, 1969, s. 2089 Guggenheim E.A.: Termodynamics, Amsterdam, North-Holland Publ.Co 1950 Burden M.H., Hunt J.D., J. Cryst. Growth, Trans., vol. 22, 1974, s. 109. Tiller W., Grain size control during ingot solidification, Trans, of the Metall. Soc., v. 224, s. 448. Witzke S., Riquet J., Durand F., Acta Metallurgica, vol.29, 1981, s. 365 Fredrikson H., Hillert M., On formation of the central equiaxed zone in ingots. Met.Trans., v. 3, 1972, s. 565 Elliot R., The formation of the equiaxed crystal zone during solidification. The British Foundryman, No.10, 1964, p. 164 Bałandin G., Formirowanije kristalliczeskowo strojenija otliwok, Maszinistr., Moskwa 1973. Flemings M., Solidification processing, Met. Trans., vol,5, 1974, s Southin R.T., Trans,TMS-AIME, v.236, 1967, s.220. Fraś E., Krzepnięcie metali i stopów, WN PWN, Warszawa 1992. Kurz W., Fisher D. J., Fundamentals solidification, Trans, Tech. Publ., Paris 1984. Chalmers B., The structure of ingots, J. Aust. Int. Met.,vol.8, 1963, s. 224. Southin R., Nucleation of equiaxed zone i cast metals, TMS-AIME, vol.229, 1967, s.220. Ohno A., The solidification of metals. Chijin Shoken, Tokyo 1980. Jackson K., Hunt J., Uhlmann D., Soward T., Trans. TMS-AIME, vol. 236, 1967, p. 149. Langenberg F.C., Prestel G., Honeycutt C.R., Trans. TMS-AIME, vol. 221, 1961, p. 993.

3
Kinetyka krzepnięcia odlewów

4
Równoważność oddziaływań fizycznych i chemicznych
obszar jednofazowy obszar dwufazowy gdzie: Gl - gradient temperatury w cieczy, v - prędkość krystalizacji. CO, CE - stężenie cieczy i dendrytów, m - nachylenie linii likwidus, D - współczynnik dyfuzji

5
Funkcję Gibbsa, funkcję stanu można przedstawić
w następującej postaci: Zapis dotyczy całego, dowolnego układu złożonego obejmującego parametry ekstensywne i intensywne. Przyjmowane założenia dotyczą zawsze ustalania jednego z nich i zmienności drugiego. Energię swobodną rozłożono na składowe: objętościową i powierzchniową, przy czym objętość przypisano sumie objętości składników przed tworzeniem stopu jak i po jego wytworzeniu wraz z powstałymi produktami reakcji między nimi. Mamy zatem dla: objętości: V=Vi+Vi-n (VA=0) energii wewnętrznej: U=UV+UA entropii: S=SV+SA energii swobodnej F=FV+FA liczby moli ni=nVi+nAi zależności:
 energii wewnętrznej: U=UV+UA. entropii: S=SV+SA. energii swobodnej F=FV+FA. liczby moli ni=nVi+nAi. zależności:")
6
W technologiach wykorzystujących dodatkowe oddziaływania fizyczne jak np.: pola elektryczne, magnetyczne konieczne jest ich uwzględnienie w ogólnym bilansie energetycznym: gdzie: S - entropia, T - temperatura, V - objętość, P - ciśnienie, Q - ładunek elektryczny, ψ- potencjał elektryczny, M - moment magnetyczny, B - indukcja magnetyczna, μi- potencjał chemiczny składnika, ni - ilość składnika i (mol), ω – energia powierzchniowa, A - powierzchnia.
, ω – energia powierzchniowa, A - powierzchnia.")
7
Wszystkie procesy dynamicznego oddziaływania na kąpiel można rozpatrywać jako wzrost energii Fermiego, a także energii wewnętrznej cieczy. Powiększa się, zatem także metastabilność układu przyspieszając w ten sposób krystalizację. Makroskopowe lub lokalne zwiększanie ciśnienia w kąpieli jest termodynamicznie analogiczne do zwiększania stopnia przechłodzenia. Znajduje to potwierdzenie w teorii W.A. Tillera dotyczącej zarodkowania w metalach i stopach a opisanych równaniem Clausiusa-Clapeyrona: gdzie: dp wzrost ciśnienia, dTE- rzeczywiste zwiększenie przechłodzenia, dHf – molowe ciepło topnienia, ΔVf -zmiana jednostkowej objętości cieczy przy krzepnięciu, L - ciepło krystalizacji

8
Wyznaczając następnie temperaturę TE i wstawiając ją do zależności na krytyczny promień zarodka homogenicznego lub heterogenicznego, w klasycznym rozumieniu uzyskuje się odpowiedni promień krytyczny (rx) powstały w nowych warunkach: gdzie: ω i Θ to odpowiednio - swobodna energia powierzchniowa między kryształem a cieczą i kąt zwilżania

9
Z energii swobodnej Helmholtza:
Korzystając z zależności na energię swobodną – funkcji Helmholtza i potencjału termodynamicznego Herringa można wykazać współzależność i równoważność oddziaływań fizycznych i chemicznych, głównie w strefie granicznej reprezentowanej przez powierzchnię kontaktu, lecz także w objętości otaczającej powierzchnię kontaktu: Z energii swobodnej Helmholtza: energia swobodna powierzchni: energia użyteczna powierzchni: gdzie: σ - napięcie powierzchniowe

10
W obszarze międzyfazowym gęstość, ciśnienie, skład chemiczny i właściwości fizyczne zmieniają się w zakresie od wartości charakterystycznych dla cieczy do wartości typowych dla kryształów. Dla zastosowań związanych z układami wielofazowymi, wskazane byłoby posługiwanie się koncepcją Guggenheima uwzględniającą niezerową grubość fazy powierzchniowej o cechach i własnościach dodatkowej fazy objętościowej. Zróżnicowanie energii powierzchniowej występuje zarówno na różnych płaszczyznach krystalograficznych tego samego metalu, ale także na różnych kierunkach krystalograficznych należących do wspólnej płaszczyzny. Jeśli przyjąć, że energia powierzchniowa jest proporcjonalna do niewysycenia wiązań atomowych wówczas minimalną energię powierzchniową będą posiadały płaszczyzny krystalograficzne gęstego upakowania atomów Lokalny dodatkowy efekt energetyczny pozostający w związku z topografią i morfologią kryształów wpływa na inicjowanie i przebieg generowania dyfuzyjnej strefy przejściowej między cieczą a kryształami. Przykładowo wielkość energii krawędziowej w atmosferze powietrza w temperaturze 900oC dla srebra wynosi: λ(100)=38 pJ/m, λ(111)=57 pJ/m. Miedź w atmosferze wodoru w temperaturze 1000oC posiada: λ(100)=20 pJ/m i λ(111)=34 pJ/m
=38 pJ/m, λ(111)=57 pJ/m. Miedź w atmosferze wodoru w temperaturze 1000oC posiada: λ(100)=20 pJ/m i λ(111)=34 pJ/m.")
11
Tillera lub Witzkego– o zarodkowaniu przed frontem krystalizacji
Dla zaistnienia zarodkowania niezbędne jest stworzenie właściwego przechłodzenia cieczy przed frontem krystalizacji. Zarodki znajdujące się w innych obszarach odlewu powstają na granicach faz - między innymi przez kontakt ciekłego metalu z elementami formy, w tym głównie układu wlewowego. Modele opisujące ten zakres dziedziny zarodkowania to między innymi: Chalmersa – tzw. „big bang” oparty na idei zarodkowania poprzez kryształy zamrożone powstałe na wewnętrznej powierzchni wnęki formy Ohno – polegający na zarodkotwórczym działaniu kryształów oderwanych od powierzchni wnęki formy Jaksona i in. – według, których powstaje w kąpieli dyspersja kryształów poprzez ich nadtapianie i rozmnażanie Southina – w którym zarodki powstają na powierzchni ciekłego metalu. Jest tzw. model deszczowy Tillera lub Witzkego– o zarodkowaniu przed frontem krystalizacji Uwzględniające zewnętrzne pola siłowe – wymuszoną konwekcję i w jej następstwie fragmentację dendrytów na skutek -fluktuacji temperatury -mechanicznego działania ciekłego metalu -połączonego wpływu czynników: kinetycznego, stężeniowego i cieplnego

12
W procesie zarodkowania, przy wspomaganiu zwilżania fizycznymi oddziaływaniami, sumaryczna powierzchnia międzyfazowa nie dość, że jest wielokrotnie większa niż w tradycyjnych stopach to jeszcze dodatkowo aktywowana może zmieniać swoją powierzchnię kontaktu w zależności od temperatury, lokalnego składu chemicznego, chwilowego stężenia i intensywności zewnętrznego pola siłowego. Podsumowując można postawić tezę: zwiększenie energii powierzchniowej jest skutkiem poprawnego zwilżania oraz właściwej adhezji i dyfuzji, powodując minimalizację naprężeń i ich homogenizację

13
Trajektoria ruchu pojedynczego jonu Fe w temperaturze 1823 K rzutowana na płaszczyznę poziomą YOX
jest on związany z ruchem dużej grupy sąsiednich jonów, co prowadzi do stabilności lokalnej struktury w czasie 6·10-3 s. Jon w tym czasie może znajdować się w stanie ruchu drgającego względem prawie nieruchomego środka równowagi, a następnie wykonuje przeskok do nowego ośrodka drgań.

14
Charakterystyki struktur atomowych metali w stanie stałym i ciekłym
Stan skupienia stały ciekły Liczba koordyna- cyjna Średnia odległość miedzy atomami l0-1nm Temperatura K koordynacyjna między atoniami 10-1nm Al 12 2,86 973 10,6 2,96 Zn 6—6 2,16—2,90 763 11 2,94 Hg 3,47—3,00 293 3,30 Na 8 3,72 373 4,38 Ge 4 2,34 1273 2,70 Bi 3—3 3,09—3,46 613 7—8 3,32

15
Z tabl. wynika, że dla czystych metali (Al, Zn, Hg, Na) liczba koordynacyjna przy roztapianiu nie zmienia się lub zmniejsza w niewielkim stopniu. Dla nich upakowanie w stanie ciekłym i stałym odpowiada jednej klasie struktur. Dla innych metali (Bi, Ge) liczba koordynacyjna przy roztapianiu wzrasta. W stanie stałym metale te mają strukturę o mniejszym stopniu upakowania (defekty struktury), a w ciekłym — bardziej gęstą, odnoszącą się już do drugiej klasy struktur. Roztapianie powoduje częściowe rozerwanie połączeń między atomami, co pozwala zapełnić atomami „pustki" krystalograficzne, istniejące w sieci ciała stałego. Wreszcie część metali, mających w stanie stałym sieć tetragonalną (Zn, Hg, Bi), traci tetragonalność po roztopieniu. Ogólnie stwierdza się, że metale po stopieniu rozszerzają się tylko o 3 - 5% (Bi nawet się kurczy), tak że liczba i upakowanie najbliższych sąsiadów nie mogą ulegać poważniejszym zmianom
, tak że liczba i upakowanie najbliższych sąsiadów nie mogą ulegać poważniejszym zmianom.")
16
Model rozłożenia atomów w cieczy (ciemne kule) i w fazie stałej (jasne kule) na powierzchni:
a) atomowogładkiej, b) szorstkiej — rozmytej; H — entalpia,----- umowna granica rozdziału faz: ciecz — faza stała
 atomowogładkiej, b) szorstkiej — rozmytej; H — entalpia,----- umowna granica rozdziału faz: ciecz — faza stała.")
17
Energia swobodna układu ΔG w funkcji zmiany rozmieszczenia atomów, spowodowanej przyłączeniem nowego atomu E1 do fluktuacji En wg J.W. Christiana ΔGA — energia stanu aktywowanego, ΔGn — energia fluktuacji n atomów, ΔGn+1 — energia fluktuacji n +1 atomów

18
Topnienie i krystalizacja:
a) w układzie atomowym, b) energetycznym; l — atom w procesie topnienia, 2 — atom w procesie krystalizacji, ΔGL, ΔGS — energia swobodna atomów w fazie ciekłej i stałej, ΔGt, ΔGkryst— energia aktywacji topnienia i krystalizacji, L1 — utajone ciepło krzepnięcia, x — odległość od frontu krzepnięcia
 w układzie atomowym, b) energetycznym; l — atom w procesie topnienia, 2 — atom w procesie krystalizacji, ΔGL, ΔGS — energia swobodna atomów w fazie ciekłej i stałej, ΔGt, ΔGkryst— energia aktywacji. topnienia i krystalizacji, L1 — utajone ciepło krzepnięcia, x — odległość od frontu krzepnięcia.")
19
Zależność energii swobodnej fazy stałej ΔGS, i ciekłej ΔGL od temperatury T; ΔGi — termodynamiczny potencjał przemiany (krystalizacji), odpowiadający przechłodzeniu ΔT, TE — temperatura równowagowa
, odpowiadający przechłodzeniu ΔT, TE — temperatura równowagowa")
20
Zmiana energii swobodnej ΔG, przy tworzeniu zarodka kulistego o promieniu r; r* — promień krytyczny zarodka, ΔGi* — krytyczna energia swobodna zarodkowania

21
r* Wpływ przechłodzenia ΔT
i energii swobodnej zarodkowania ΔG na promień krytyczny zarodka r*: a) zależność termodynamicznego potencjału zarodkowania ΔGi od przechłodzenia ΔT1; b) zależność promienia zarodka krytycznego od krytycznej energii swobodnej zarodkowania ΔGi*i i temperatury T; c) zależność wielkości zarodka krytycznego od przechłodzenia ΔT r*
 zależność termodynamicznego potencjału zarodkowania ΔGi od przechłodzenia ΔT1; b) zależność promienia zarodka krytycznego od krytycznej energii swobodnej zarodkowania ΔGi*i i temperatury T; c) zależność wielkości zarodka krytycznego od przechłodzenia ΔT. r*")
22
Lit 933 544 600 505 Gal 303 Sód 504 459 Metal Temperatura K Nap. Pow.
N/m 10-3 N/m Aluminium 933 860±20 Molibden 2893 2240 Antymon 900,5 395 ±20 Nikiel 1823 1924 Bizmut 544 380±10 Ołów 600 Chrom 1888 1420 Platyna 2273 1819 Cyna 505 525 ±10 Potas 373 573; 773 98 75; 54 Cynk 692,5 750±20 Rtęć 234,63 ; 273 473 573 623 500+15 470 450 430 410 400 Cyrkon 2173 1380 Gal 303 725 ±10 Iryd 2623 2310 Sód 373; 475 723 190; 180 171 156 Kadm 504 550±10 Kobalt 1768 1936 1740 Srebro 1233,5 923 Krzem 1723 725 ±800 Srebro w fazie stałej 1040 1235 Lit 459 Tytan 1998 1325 Magnez 520±10 Uran 1423 1020 Mangan 1533 1050 Wanad 1983 1710 Miedź 1473 1300 Wapń 1083 255 Miedź w fazie stałej 1100 1852 Wolfram 3653 2680 Złoto 1336 1128 Złoto w fazie stałej 1260 1610 Żelazo 1835

23
Schemat zarodka: a) homogenicznego, b), c), heterogenicznego, d) katalitycznego; L — faza ciekła, S — faza stała, P — podłoże, W — warstwa przejściowa, V*,r* — objętość i promień zarodka krytycznego

25
—_ciecz (L) Zarodkowanie heterogeniczne (na podłożu P); r*, V* — promień i objętość zarodka homogenicznego, rp*, Vp* — promień i objętość zarodka heterogenicznego (wycinka kulistego o wysokości h sprowadzonego do kuli)
; r*, V* — promień i objętość zarodka homogenicznego, rp*, Vp* — promień i objętość zarodka heterogenicznego (wycinka kulistego o wysokości h sprowadzonego do kuli)")
26
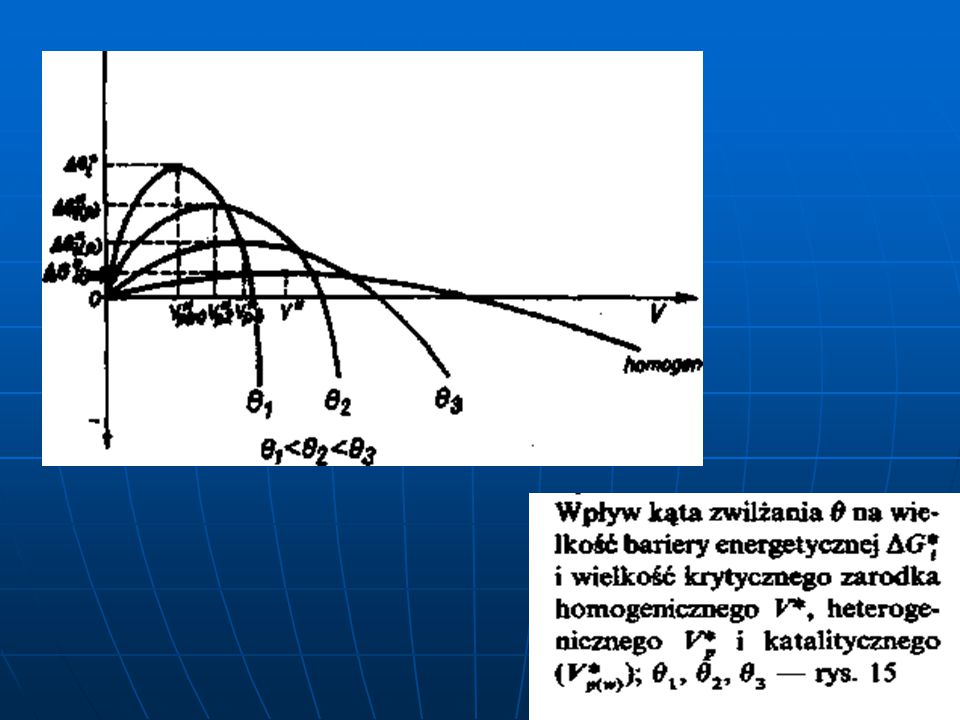
Zależność między stosunkiem objętości zarodka heterogenicznego V
Zależność między stosunkiem objętości zarodka heterogenicznego V*p i homogenicznego V* a krawędziowym kątem zwilżania Θ

27
Schemat warunków zarodkowania heterogenicznego w funkcji przechłodzenia ΔT i krawędziowego kąta zwilżania

28
Energetyczna nadrzędność heterogenicznego zarodkowania jest tak znaczna, iż nawet w czystych metalach przeważa pierwszeństwo w tworzeniu zarodków heterogenicznych. Miarą tych różnic może być wielkość przechłodzenia w stosunku do zarodkowania homogenicznego. W badaniach eksperymentalnych, gdzie z założenia dąży się do minimalizacji badanych objętości, chociażby dla zmniejszenia prawdopodobieństwa oddziaływań wtrąceń mogących być podkładką zarodkotwórczą, różnice w wielkości przechłodzenia mogą wynosić kilkadziesiąt stopni. Dla przykładu podstawiając do wzoru na wielkość krytyczną zarodka: można wyznaczyć promień r* jako funkcję wielkości przechłodzenia. Przykładowa zależność dla miedzi:

29
Zależność wielkości promienia krytycznego zarodka miedzi w zależności od stopnia przechłodzenia ΔT
Podstawą analiz powinny być ze względu na procesy zarodkowania wielkości rzędu: {kilkudziesięciu ÷ kilkuset [Å]}. Wynika to z wielkości zarodków rzędu kilku – kilkudziesięciu [nm]. Na rysunku poniżej zakreskowany obszar oznacza intensywne zarodkowanie dla kątów od 0÷25o na płaszczyźnie, dla kątów 25÷45o dla progów, krawędzi oraz 45÷90o w szczelinach oraz 90÷120o w we wgłębieniach 90÷120o. Przy czym w większości przypadków głębokość defektów powierzchniowych jest rzędu ok. 100 [Å]

30
zarodkowania heterogenicznego
Porównanie energii stanu aktywowanego, niezbędnej do zarodkowania homogenicznego i heterogenicznego w zależności od nierówności na powierzchni rozdziału fazy stałej i ciekłej wg W. Missola

31
a) wklęsłej, b) płaskiej c) wypukłej
Tworzenie kryształów na powierzchni podłoża: a) wklęsłej, b) płaskiej c) wypukłej
 wklęsłej, b) płaskiej. c) wypukłej.")
32
Przykładowe tworzenie jamek trawienia cieplnego na płaszczyznach (111) i (100) polikrystalicznego srebra (40 h, 700°C, PH2O/PH2 = 0,08)
 i (100) polikrystalicznego srebra (40 h, 700°C, PH2O/PH2 = 0,08)")
33
Kształt powierzchni międzyfazowej ciecz metaliczna – kryształ zależy m
Kształt powierzchni międzyfazowej ciecz metaliczna – kryształ zależy m. in. od gradientu temperatury w pobliżu frontu krystalizacji. Tiller dokonał szacunkowej oceny wpływu dyfuzji składnika oraz transportu ciepła [81]. Odległość, na którą następuje dyfuzja w jednostce czasu przy jednostkowym gradiencie stężenia jest około sto razy mniejsza niż droga przemieszczenia frontu krystalizacji. Natomiast odległość, na którą przenoszone jest ciepło w jednostce czasu przy jednostkowym gradiencie temperatury jest ok. sto razy większa niż wielkość przemieszczenia frontu krystalizacji. Powierzchnia międzyfazowa, która szybciej reaguje na zmianę warunków termodynamicznych dominuje w kształtowaniu struktury Rozwinięta powierzchnia międzyfazowa może być definiowana za pomocą promienia krzywizny. Może być rozpatrywana w kategoriach przyrostu temperatury lub ciśnienia lub też zmiany stężenia – głównych czynników krystalizacyjnych. Jeśli w analizowanej przestrzeni napięcie powierzchniowe σ jest izotropowe wówczas przyrost energii swobodnej spowodowanej obecnością powierzchni zakrzywionej o promieniu r wynosi: gdzie: to uśredniona krzywizna

34
ΔGρ – zmiana energii swobodnej wywołana krzywizną
ρ1 ρ2, - zorientowane promienie główne prostopadłych krzywizn powierzchni kryształu. Klasyczna postać równania Gibbsa-Thomsona pokazuje zależność równowagowej temperatury krystalizacji powierzchni płaskiej Te i zakrzywionej Tρ frontu krystalizacji. Zależność odnosi się zasadniczo do czystych metali. Zakłada się równość równowagowych współczynników rozdziału składnika między cieczą i kryształem zakrzywionym i płaskim. Jeśli przyjąć, że promienie r prostopadłe krzywizn są równe, co odpowiada powierzchni sferycznej wówczas przyrost temperatury zależąc od promienia krzywizny r jest dwa razy wyższy niż w przypadku powierzchni jednokrotnie zakrzywionej – np. walcowej. Przykład wpływu obu przypadków wskazuje na skalę oddziaływań. Wzrost rozwinięcia powierzchni heterogenicznego zarodkowania a tym samym wzrost powierzchni frontu wzrostu kryształów oraz minimalizacja promienia krzywizn celowo kształtowanych powierzchniowo komponentów zbrojących, prowadzi do zmniejszania równowagowej temperatury krystalizacji.

35
Promień krzywizny powierzchni międzyfazowej ciecz-ciało stałe można wyznaczyć korzystając z warunku
gdzie: Tx – temperatura powierzchni rozdziału ciecz- ciało stałe, - gradient temperatury, Δx – przemieszczenie względem izotermy Te; Te równowagowa temperatura krystalizacji czystego metalu Po podstawieniu:

36
Rozpatrując prosty przypadek równowagi sił powierzchniowych dwufazowej osnowy i cząstki zbrojącej spełniony musi być warunek Neumana, równości napięć powierzchniowych: Równowaga granic rozdziału w układzie trójfazowym

37
Granice między ziarnami tej samej fazy mają jednakowe energie powierzchniowe, natomiast granice między ziarnami różnych faz mają inne wartości energii powierzchniowych i inne napięcia powierzchniowe Równowagowy dwuścienny kąt zwilżania Θ2 na styku dwóch ziarn jednej fazy z drugą fazą

38
Uzyskanie płaskich ścian jest możliwe tylko wówczas, gdy układ ma zrównoważone nadmiary powierzchniowe energii, czyli jest stabilny. Kąty tetraedryczne dążą do wartości 109,5 o. Warunki związane z wypełnieniem przestrzeni i równowagi napięć powierzchniowych spełniają ziarna w postaci czternastościanów

39
Ułożenie i kształt ziarn wypełniających przestrzeń w pełni przy zachowaniu płaskich ścian:
model czternastościany oraz powierzchnia odpowiadającego modelowi zgładu

40
Przy krystalizacji pojedynczych kryształów równowagowy kształt odpowiada minimalnej energii powierzchniowej przy stałej objętości. Wynika to z prawa Gibbsa: Kształt kulisty jest uprzywilejowany z założenia w warunkach izotropowego pola napięcia powierzchniowego. Przy anizotropii napięcia powierzchniowego spowodowanego odmiennością energii powierzchniowych w zależności od orientacji krystalograficznej na powierzchnia kryształu wystąpią granice o niesferycznych kształtach. Kształt ziarna wg Wulfa pokazany na rysunku powiązany z energiami i napięciami powierzchniowymi:

41
Kryształ Wulfa gdzie: F, V, odpowiednio powierzchnia całkowita , objętość kryształu, b, d – stałe zależne od kształtu, σi napięcie powierzchniowe na ścianie przy wysokości hi zgodnie z rysunkiem

Podobne prezentacje