Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Poznanie genomu człowieka (wg. artykułów z Science i Nature)
Jerzy Tiuryn Instytut Informatyki Uniwersytet Warszawski

3
Dwa artykuły „Initial sequencing and analysis of the human genome”, International Human Genome Sequencing Consortium, Nature, , 2001 ( ). „The sequence of the human genome”, J.C. Venter, et.al., Science, ( ).
. „The sequence of the human genome , J.C. Venter, et.al., Science, ( ).")
4
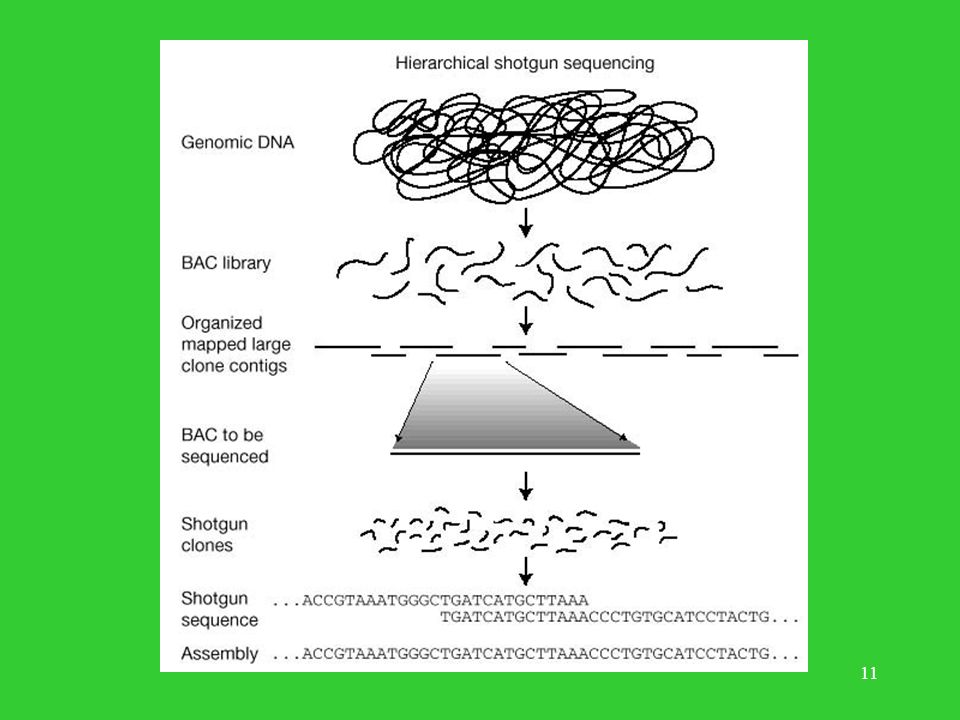
Plan wykładu Historia poznania genomu człowieka.
Metoda konsorcjum (hierarchiczne sekwencjonowanie metodą ‘shotgun’). Metoda Ventera ‘whole-genome shotgun approach’. Co wiadomo o liczbie genów w genomie człowieka? Porównanie obu metod.
. Metoda Ventera ‘whole-genome shotgun approach’. Co wiadomo o liczbie genów w genomie człowieka Porównanie obu metod.")
5
Historia poznania genomu człowieka
1953, James Watson, Francis Crick, : struktura DNA.

6
1977, F. Sanger (metoda dideoxy), 500-750bp.
1977, F. Sanger: zsekewncjonowanie pierwszego ludzkiego genu. , genomy bakteryjnych wirusów (φX174, Lambda), genom wirusa zwierzęcego SV40, ludzkie mitochondrium. 1985, K. Mullis: technika PCR. 1987, D. Burke, M. Olson, G. Carle: YAC. 1989, Olson, Hood, Botstein, Cantor: strategia mapowania przy użyciu STS.
, genom wirusa zwierzęcego SV40, ludzkie mitochondrium. 1985, K. Mullis: technika PCR. 1987, D. Burke, M. Olson, G. Carle: YAC. 1989, Olson, Hood, Botstein, Cantor: strategia mapowania przy użyciu STS.")
7
1995, J. C. Venter (Heamophilus influenzae) 1
1995, J.C. Venter (Heamophilus influenzae) 1.8 Mb, metoda ‘whole-genome shotgun sequencing’. 1996, Międzynarodowe konsorcjum (Saccharomyces cerevisiae) 13.5 Mb. 1997, Blattner, Plunkett (Escherichia coli) 5 Mb. 1998, Venter: założenie firmy Celera Genomics (deklaracja: sekwencja genomu człowieka w 3 lata, za 300 M$).
 1.8 Mb, metoda ‘whole-genome shotgun sequencing’. 1996, Międzynarodowe konsorcjum (Saccharomyces cerevisiae) 13.5 Mb. 1997, Blattner, Plunkett (Escherichia coli) 5 Mb. 1998, Venter: założenie firmy Celera Genomics (deklaracja: sekwencja genomu człowieka w 3 lata, za 300 M$).")
8
1998, Sulston, Waterson (Caenorhabditis elegans) 100 Mb.
1999, GB, Japonia, USA: chromosom nr.22, 35 Mb. 2000, Venter (Drosophila melanogaster) 120 Mb, testowanie metody WGSS dla niezbyt dużego genomu. 2000, Niemcy, Japonia: chromosom nr. 21, 34 Mb. 2000, Międzynarodowe Konsorcjum (Arabidopsis thaliana), 100 Mb. 2001, HGP i Celera publikują draft genomu człowieka, 3.3Gb.
 120 Mb, testowanie metody WGSS dla niezbyt dużego genomu. 2000, Niemcy, Japonia: chromosom nr. 21, 34 Mb. 2000, Międzynarodowe Konsorcjum (Arabidopsis thaliana), 100 Mb. 2001, HGP i Celera publikują draft genomu człowieka, 3.3Gb.")
9
Główne trudności w sekwencjonowaniu genomu człowieka
Rozmiar genomu (~3Gb). Duża część genomu zawiera repetytywne fragmenty. Przykładowo część genomu zawierająca repetytywne fragmenty dla różnych organizmów: Bakterie: ~1.5% Muszka owocowa: ~3% Człowiek: >50%
. Duża część genomu zawiera repetytywne fragmenty. Przykładowo część genomu zawierająca repetytywne fragmenty dla różnych organizmów: Bakterie: ~1.5% Muszka owocowa: ~3% Człowiek: >50%")
10
Metoda Konsorcjum map-based, BAC-based, clone-by-clone
Pozyskiwanie materiału genetycznego. Budowa mapy fizycznej genomu w oparciu o klony. Trawienie poszczególnych klonów enzymami restrykcyjnymi – ‘odcisk palca’. Budowa kontigów i przypisanie ich do miejsc na chromosomach (STS). Wybór klonów z kontigów do sekwencjonowania. Sekwencjonowanie metodą ‘shotgun’ wybranych klonów. Składanie genomu.
. Wybór klonów z kontigów do sekwencjonowania. Sekwencjonowanie metodą ‘shotgun’ wybranych klonów. Składanie genomu.")
12
Pozyskiwanie materiału genetycznego
Ochotnicy (różne środowiska etniczne), ‘kto pierwszy ten lepszy’. Samplig laboratory: usunięcie identyfikatorów, nadanie losowych oznaczeń, przesłanie do processing lab. Processing laboratory: usuwa wszystkie oznaczenia i zmienia je na inne, niszczy dokumentację oznaczeń, wybiera losowo 5-10 próbek do dalszej analizy.
, ‘kto pierwszy ten lepszy’. Samplig laboratory: usunięcie identyfikatorów, nadanie losowych oznaczeń, przesłanie do processing lab. Processing laboratory: usuwa wszystkie oznaczenia i zmienia je na inne, niszczy dokumentację oznaczeń, wybiera losowo 5-10 próbek do dalszej analizy.")
13
Linia produkcyjna do przygotowywania próbek
Whitehead Institute, Center for Genome Research

14
Klony Plazmidy (~ 4Kb). Kosmidy (~ 40Kb).
Yeast Artificial Chromosome, YAC (do 500Kb). Bacterial Artificial Chromosome, BAC ( Kb).
. Bacterial Artificial Chromosome, BAC ( Kb).")
15
Mapa fizyczna Biblioteki klonów zbudowane z materiału genetycznego. ( klonów BAC lub PAC, 65-krotne pokrycie genomu). Każdy klon rozmiaru Kb. Wybrano ~ klonów do budowy mapy fizycznej. (20 krotne pokrycie genomu). Każdy klon poddano trawieniu enzymem restrykcyjnym i zmierzono rozmiary fragmentów przy pomocy elektroforezy na żelu z agarozy. Tak powstaje linia papilarna (fingerprint) klonu. Linie papilarne są użyte do identyfikacji klonów i do szacowania wielkości nałożenia jednego klonu na drugi.
. Każdy klon rozmiaru Kb. Wybrano ~ klonów do budowy mapy fizycznej. (20 krotne pokrycie genomu). Każdy klon poddano trawieniu enzymem restrykcyjnym i zmierzono rozmiary fragmentów przy pomocy elektroforezy na żelu z agarozy. Tak powstaje linia papilarna (fingerprint) klonu. Linie papilarne są użyte do identyfikacji klonów i do szacowania wielkości nałożenia jednego klonu na drugi.")
16
Mapa fizyczna, c.d. Linie papilarne klonów zostały użyte do budowy tzw. kontigów (nakładające się na siebie spójne fragmenty utworzone z klonów). Kontigi zostały przyporządkowane miejscom na chromosomach przy pomocy znaczników STS (STS = Sequence Tagged Site ~ 500bp, jednoznaczna sekwencja na chromosomie, dla której są znane primery PCR).
.")
17
Przykład dwóch kontigów

18
Faza sekwencjonowania
Wybór klonów z kontigów, tak aby uzyskać pokrycie genomu (aby przyspieszyć proces, zrezygnowano z poszukiwania minimalnego pokrycia). Wybrano ~ klonów.
. Wybrano ~ klonów.")
19
Faza sekwencjonowania: każdy klon metodą ‘shotgun’
Klon powiela się w wielu kopiach. Wszystkie kopie tnie się na małe kawałki (enzymy restrykcyjne) ‘losowo’. Porządek i orientacja kawałków są tracone. Wybiera się losowo dostatecznie dużo kawałków (5-10 krotne pokrycie, zgodnie z formułą Landera/Watermana) i dla każdego kawałka sekwencjonuje się prefiks o długości ~ 500bp. Powstają tzw. czyste odczyty.
 ‘losowo’. Porządek i orientacja kawałków są tracone. Wybiera się losowo dostatecznie dużo kawałków (5-10 krotne pokrycie, zgodnie z formułą Landera/Watermana) i dla każdego kawałka sekwencjonuje się prefiks o długości ~ 500bp. Powstają tzw. czyste odczyty.")
20
Uwagi na temat metody ‘shotgun’
W praktyce wybór fragmentów nie jest jednorodny (powody molekularno-biologiczne, a nie probabilistyczne). To powoduje powstawanie dziur w odczytywanej sekwencji. Są dwa stopnie jakości metody ‘shotgun’: ‘half-shotgun’ 4-5 krotne pokrycie, w wyniku mamy draft genomu. ‘full-shotgun’ 8-10 krotne pokrycie, w wyniku mamy podstawę do dokładnego opisu genomu.
. To powoduje powstawanie dziur w odczytywanej sekwencji. Są dwa stopnie jakości metody ‘shotgun’: ‘half-shotgun’ 4-5 krotne pokrycie, w wyniku mamy draft genomu. ‘full-shotgun’ 8-10 krotne pokrycie, w wyniku mamy podstawę do dokładnego opisu genomu.")
21
Uzyskano 23Gb danych w czystych odczytach.
Niektóre centra osiągnęły wydajność reakcji sekwencjonowania na 12 godzin. Wydajność wszystkich centrów osiągnięta w czerwcu 2000: 1 pokrycie genomu na 6 tygodni (1Kb/sek. przez 24h/dobę, cały czas). Każdy nukleotyd był odczytany średnio 4.5 raza.
. Każdy nukleotyd był odczytany średnio 4.5 raza.")
22
w postaci finalnej było 835Mb sekwencji genomu (wliczając chromosomy 21 i 22). Na koniec roku 2000 było ~ 1Gb sekwencji w finalnej postaci (finalna postać = prawdopodobieństwo błędu odczytu nukleotydu < 1/10.000, żadnych dziur)
.")
23
Składanie sekwencji (1)
Analiza nałożeń (overlap detection): dane dwa słowa W,V, znajdź sufiks w W oraz prefiks w V o maksymalnym podobieństwie (w sensie uliniowienia; mogą być wstawiane spacje). Jest to problem natury algorytmicznej. Dane o nałożeniach przechowujemy.
: dane dwa słowa W,V, znajdź sufiks w W oraz prefiks w V o maksymalnym podobieństwie (w sensie uliniowienia; mogą być wstawiane spacje). Jest to problem natury algorytmicznej. Dane o nałożeniach przechowujemy.")
24
Składanie sekwencji (2)
Ułożenie podsłów (substring layout). Zachłanny algorytm: znajdź parę słów o maksymalnym podobieństwie sufiks/prefiks. Później następną parę. Albo powstają dwa kontigi, albo jeden o trzech słowach. Podobne do wielokrotnego uliniowienia. Dodawanie nowych par powoduje wstawianie spacji (rozsuwanie). W ten sposób powstają kontigi nakrywające większość odtwarzanej sekwencji.
. Zachłanny algorytm: znajdź parę słów o maksymalnym podobieństwie sufiks/prefiks. Później następną parę. Albo powstają dwa kontigi, albo jeden o trzech słowach. Podobne do wielokrotnego uliniowienia. Dodawanie nowych par powoduje wstawianie spacji (rozsuwanie). W ten sposób powstają kontigi nakrywające większość odtwarzanej sekwencji.")
25
Składanie sekwencji (3)
Decydowanie konsensusu: uzgodnienie jaka litera ma stać na danej pozycji w kontigu. Stosowane są różne podejścia, często metoda większościowa (tu są subtelne problemy). W projekcie średnie pokrycie klonu kontigami wynosiło 96%, a średnie przerwy pomiędzy kontigami miały ~ 500bp.
. W projekcie średnie pokrycie klonu kontigami wynosiło 96%, a średnie przerwy pomiędzy kontigami miały ~ 500bp.")
26
Dwa rodzaje kontigów Kontigi pochodzące z jednego klonu.
Mega-kontigi pochodzące z analizy linii papilarnych poszczególnych klonów.

27
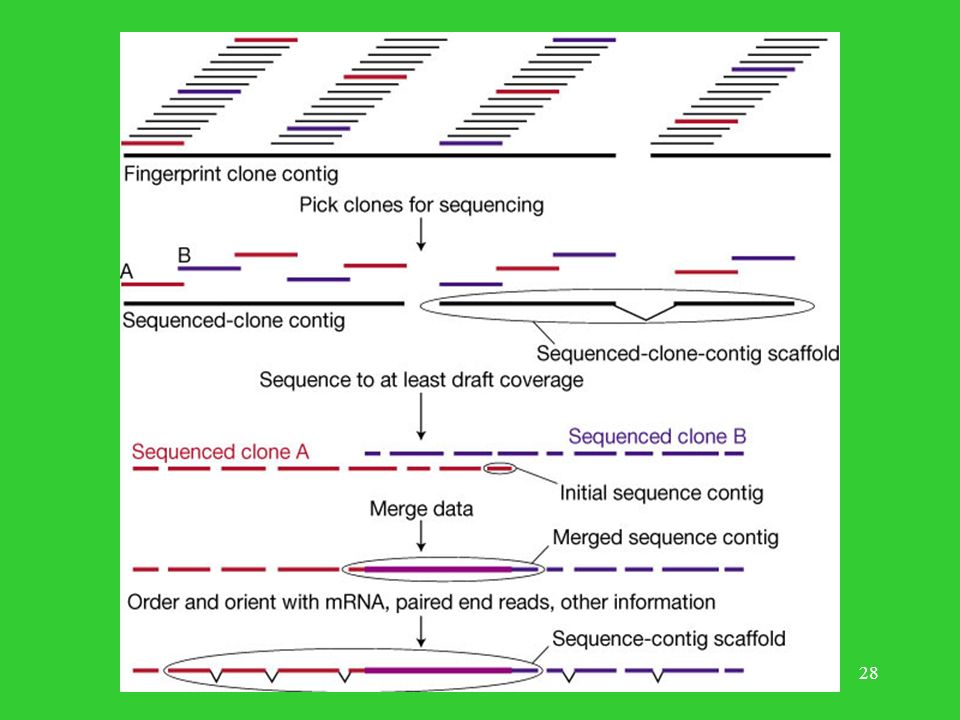
Logistyka składania genomu
Składanie pojedynczych klonów. Związanie zsekwencjonowanych klonów z pozycjami na fizycznej mapie genomu. Poprawianie niezgodności.

29
Kroki w procesie składania genomu z kontigów pochodzących z klonów A i B.

30
Jakość draftu genomu zsekwencjonowanego przez konsorcjum
Użyto oprogramowanie PHRAP (program przypisuje każdemu nukleotydowi prawdopodobieństwo błędu). 91% sekwencji ma błąd < 1/ 96% sekwencji ma błąd < 1/1.000 Są przerwy w sekwencji.
. 91% sekwencji ma błąd < 1/ % sekwencji ma błąd < 1/ Są przerwy w sekwencji.")
31
Przerwy w sekwencji (3 rodzaje)
Pomiędzy kontigami w poszczególnych klonach: łącznie 2-4% genomu jest zawarte w takich przerwach (~80Mb). Tych przerw jest ~ Pomiędzy klonami w mega-kontigach: 5% genomu (~150Mb). Jest ich ~4.000. Pomiędzy mega-kontigami (szacowanie na podstawie chr. 21 i 22) ~4% genomu.
. Tych przerw jest ~ Pomiędzy klonami w mega-kontigach: 5% genomu (~150Mb). Jest ich ~ Pomiędzy mega-kontigami (szacowanie na podstawie chr. 21 i 22) ~4% genomu.")
32
Co wiadomo na temat liczby genów?
W małych genomach geny są ściśle związane z ORFami (ORF = Open Reading Frame). U człowieka średnia długość eksonu ~145bp, natomiast introny są długie (średnio ~3300bp, ale zdarzają się introny długości > 10Kb). Przykładowo: introny (średnio) u robaka (267bp), u muchy (487bp).
. U człowieka średnia długość eksonu ~145bp, natomiast introny są długie (średnio ~3300bp, ale zdarzają się introny długości > 10Kb). Przykładowo: introny (średnio) u robaka (267bp), u muchy (487bp).")
33
Geny RNA (nie-kodujące)
Takie jak tRNA, rRNA, itd. Nie mają ORFów. Są małe i nie zawierają ogonów poly(A). Trudne do odróżnienia od pseudogenów. Łącznie znaleziono w drafcie ~700 genów RNA.
. Trudne do odróżnienia od pseudogenów. Łącznie znaleziono w drafcie ~700 genów RNA.")
34
Przykład Klasyczne (podręcznikowe) oszacowanie liczby genów tRNA u człowieka to 1310, ale ... okazało się, że jest ich w drafcie genomu tylko 497.

35
Dla innych organizmów liczba genów tRNA wynosi:

36
Geny kodujące białka Znanych jest obecnie nieco ponad sekwencji mRNA w bazie RefSeq (część bazy GenBank). Zrobiono uliniowienie z draftem genomu. Nieco ponad dało się (przynajmniej częściowo) uliniowić. 16% sekwencji mRNA wykazało podobieństwo do więcej niż jednego wystąpienia w drafcie genomu (paralogi, pseudogeny).
. Zrobiono uliniowienie z draftem genomu. Nieco ponad dało się (przynajmniej częściowo) uliniowić. 16% sekwencji mRNA wykazało podobieństwo do więcej niż jednego wystąpienia w drafcie genomu (paralogi, pseudogeny).")
37
Geny kodujące białka (rozmiary)
Duży rozrzut w rozmiarach genów (eksony i introny) człowieka. Wiele jest dłuższych niż 100Kb (rekordzista: gen dystrofiny (DMD) ma 2.4Mb. Długość kodującej sekwencji też podlega dużym wahaniom. Np. gen titiny (najdłuższa obecnie znana długość kodującej sekwencji) ma bp, liczba eksonów 178, najdłuższy ekson bp.
 człowieka. Wiele jest dłuższych niż 100Kb (rekordzista: gen dystrofiny (DMD) ma 2.4Mb. Długość kodującej sekwencji też podlega dużym wahaniom. Np. gen titiny (najdłuższa obecnie znana długość kodującej sekwencji) ma bp, liczba eksonów 178, najdłuższy ekson bp.")
38
Trudności w znajdowaniu genów w genomie człowieka
Mały iloraz sygnał/szum w genach człowieka w związku z krótkimi eksonami i bardzo długimi intronami. Ponadto kodujące sekwencje stanowią bardzo małą część genomu. Tak nie jest w drożdżach, robaku i muszce. Znając nawet dokładnie genom (tak jak to jest dla chr. 21 i 22) nadal będzie bardzo trudno odkrywać geny ‘ab initio’ .
 nadal będzie bardzo trudno odkrywać geny ‘ab initio’ .")
39
Przewidywanie liczby genów (1)
W latach 80-tych Gilbert zasugerował, że może być ~ genów w genomie człowieka. Jest to tzw. rachunek ‘back-of-the-envelope’ Typowy gen ma rozmiar ~30.000bp, rozmiar genomu jest ~3Gb, więc otrzymujemy ~ genów. Analiza na podstawie szacunku liczby wysp CpG oraz częstości związków z genami dała ~ genów.

40
Przewidywanie liczby genów (2)
Szacunki oparte o EST (EST = Expressed Sequence Tags) dawały rozrzut liczby genów w granicach
 dawały rozrzut liczby genów w granicach")
41
Obecnie stosowane metody znajdowania genów
Wystąpienie znanego EST lub mRNA. Sekwencyjne podobieństwo do znanych genów lub białek. Ab initio metoda oparta na ukrytych modelach Markowa (HMM) – używają one statystycznej informacji na temat miejsc splicingu, kodowego odchylenia (coding bias), długości eksonów i intronów (Genscan, Genie, FGENES).
 – używają one statystycznej informacji na temat miejsc splicingu, kodowego odchylenia (coding bias), długości eksonów i intronów (Genscan, Genie, FGENES).")
42
Skuteczność metod ab initio
Szacuje się, że dla muchy pojedyncze eksony mogą być odgadywane poprawnie z prawdopodobieństwem 90%, ale wszystkie eksony danego genu tylko z prawdopodobieństwem 40%. Dla człowieka podobne liczby wynoszą: 70% i 20%. Niektórzy uważają też, że w/w liczby są zbyt optymistyczne...

43
Initial Gene Index (IGI)
System Ensembl (używa Genscan, weryfikuje w oparciu o podobieństwo do białek, mRNA, EST i białkowych motywów (zawarte w bazie Pfam) dla wszystkich organizmów). System ten wygenerował predykcji genów oraz transkryptów. Po wykonaniu pewnej redukcji fragmentacji otrzymano predykcji genów. To stanowi podstawę do pierwszej wersji IGI.
 dla wszystkich organizmów). System ten wygenerował predykcji genów oraz transkryptów. Po wykonaniu pewnej redukcji fragmentacji otrzymano predykcji genów. To stanowi podstawę do pierwszej wersji IGI.")
44
Initial Gene Index (IGI)
W IGI jest znanych genów i predykcji nowych genów. Przyjmuje się, że bardziej realna liczba genów w IGI to genów (20% błędnych predykcji lub pseudogenów, 1.4 współczynnik fragmentacji). Przyjmując, że predykcje genów zawierają 60% wcześniej nieznanych genów, można oszacować łączną liczbę genów człowieka na ~
. Przyjmując, że predykcje genów zawierają 60% wcześniej nieznanych genów, można oszacować łączną liczbę genów człowieka na ~")
45
Końcowe uwagi na temat liczby genów człowieka
Obecne szacunki liczby genów oparte na próbkowaniu dają przedział Jeśli w genomie człowieka jest genów i średnia długość kodującej sekwencji wynosi 1.400bp oraz średnia długość całego genu wynosi 30Kb, to 1.5% całego genomu zajmują sekwencje kodujące, a 30% zajmują geny.

46
Końcowe uwagi na temat liczby genów człowieka
Wydaje się, że człowiek ma dwa razy więcej genów niż robak lub mucha. Geny człowieka są bardziej rozciągnięte po genomie i są one używane do budowy większej liczby alternatywnych transkryptów. Łącznie, być może, człowiek wytwarza 5 razy więcej białkowych produktów niż robak czy mucha.

47
Jaka jest naprawdę liczba genów u człowieka ...?
Michael Zhang ze współpracownikami (Cold Spring Harbour Laboratory): opracowali program First Exon Finder (grudzień 2001, Nature Genetics). Program ten wyszukuje odcinki zawierające nie-kodujące pierwsze eksony oraz sekwencje promotorowe genów. Program poprawnie zlokalizował 90% genów w zsekwencjonowanych chromosomach 21 i 22. First Exon Finder wytypował 68,000 genów w genomie człowieka. Autorzy szacują, że całkowita liczba genów w genomie człowieka waha się w granicach 50,000-60,000. Co będzie dalej ... ?
: opracowali program First Exon Finder. (grudzień 2001, Nature Genetics). Program ten wyszukuje. odcinki zawierające nie-kodujące pierwsze eksony oraz sekwencje. promotorowe genów. Program poprawnie zlokalizował 90% genów w zsekwencjonowanych chromosomach 21 i 22. First Exon Finder wytypował 68,000 genów w genomie. człowieka. Autorzy szacują, że całkowita liczba genów w. genomie człowieka waha się w granicach 50,000-60,000. Co będzie dalej ...")
48
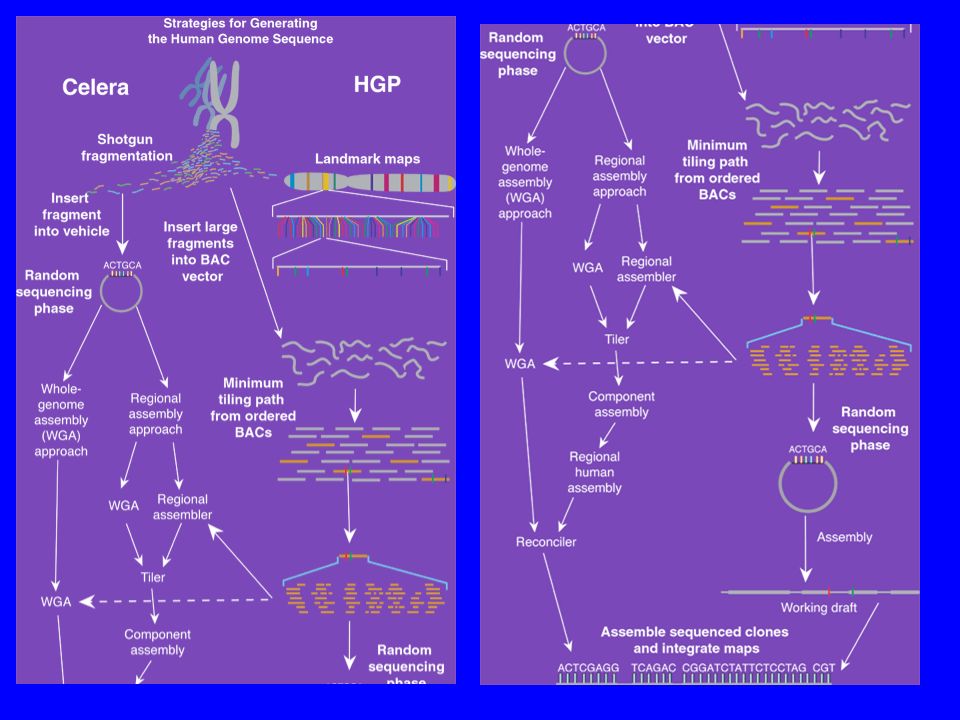
Metoda firmy Celera Genomics sekwencjonowania genomu

49
Plan Kontigi i rusztowania.
Dwie strategie asemblacji genomu (WGA, CSA). Poszukiwanie genów. Analiza genomu. Porównanie sekwencji Konsorcjum i Celery.
. Poszukiwanie genów. Analiza genomu. Porównanie sekwencji Konsorcjum i Celery.")
50
Celera 3,000 m.kw. 175,000 reakcji sekwencjonowania na dzień.
Wirtualna Farma Obliczeniowa (Compaq Alpha): 440 CPU (EV6 (400MHz), EV67(667MHz)). Każdy 2-8GB RAM. 100TB HD.
: 440 CPU (EV6 (400MHz), EV67(667MHz)). Każdy 2-8GB RAM. 100TB HD.")
51
Dane do obróbki Biblioteka plazmidów (rozmiarów 2Kb, 10Kb, 50Kb).
Konstrukcja stowarzyszonych par (mate pairs) – sekwencje bp, z każdego końca sekwencji z biblioteki plazmidów (27.27 milionów odczytów). Kontigi zbudowane z BAC’ów dostępnych z publicznych danych Konsorcjum (4.4Gb).
 – sekwencje bp, z każdego końca sekwencji z biblioteki plazmidów (27.27 milionów odczytów). Kontigi zbudowane z BAC’ów dostępnych z publicznych danych Konsorcjum (4.4Gb).")
52
Kontigi, rusztowania i stowarzyszone pary

54
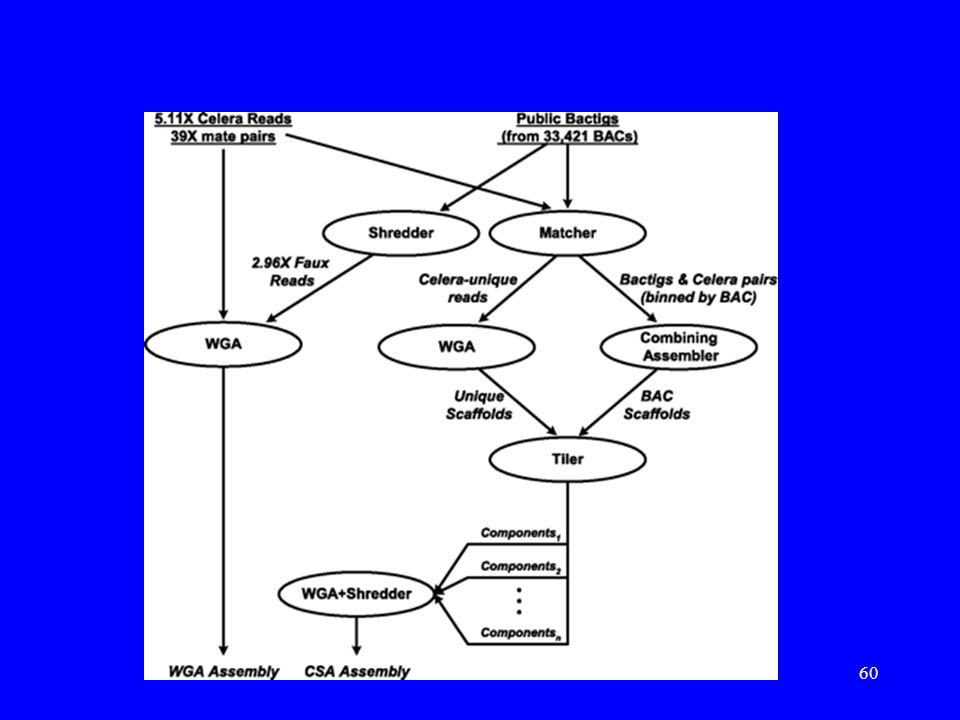
Dwie strategie asemblacji genomu
Whole-genome assembly (WGA). Compartmentalized shotgun assembly (CSA).
. Compartmentalized shotgun assembly (CSA).")
55
Asemblacja WGA Analiza nakryć (overlaps) – 10,000h czasu CPU, 40 komputerów (4-procesorowy Alpha), 4GB RAM każdy. Równoległość. Wybór jednoznacznych kontigów (unitigi) – 73.6% genomu. Wykorzystanie par stowarzyszonych do budowy rusztowań (scaffolds). Uzupełnianie dziur w rusztowaniach (fazy ‘rocks’ oraz ‘stones’).
 – 73.6% genomu. Wykorzystanie par stowarzyszonych do budowy rusztowań (scaffolds). Uzupełnianie dziur w rusztowaniach (fazy ‘rocks’ oraz ‘stones’).")
56
Asemblacja CSA (Matcher): Rozdzielenie danych Celery na te, które pasują do BAC’ów z danych publicznych i na resztę (21 milionów odczytów pasowało, a 3 miliony były nowe).
: Rozdzielenie danych Celery na te, które pasują do BAC’ów z danych publicznych i na resztę (21 milionów odczytów pasowało, a 3 miliony były nowe).")
57
Asemblacja CSA, c.d. (Combining Assembler): Dla tych z pierwszej grupy, dla każdego BAC’a wzięto kontigi z HGP oraz pasujące odczyty Celery. Użyto WGA do zbudowania rusztowań (zwykle 1 lub 2) pokrywających w ~95% ten BAC. Asemblacja wysokiej jakości.
 pokrywających w ~95% ten BAC. Asemblacja wysokiej jakości.")
58
Asemblacja CSA, c.d. (WGA): Dla drugiej grupy (nowe dane) przeprowadzono WGA. (Tiler): Analiza porządku i nakryć dla rusztowań pochodzących z BAC’ów i z rusztowań zbudowanych dla nowych danych. Użyto: pary stowarzyszone dla klonów 50Kb i dla BAC’ów oraz markery STS. Powstało w ten sposób składowych (components) obejmujących ~2.92Gb.
: Analiza porządku i nakryć dla rusztowań pochodzących z BAC’ów i z rusztowań zbudowanych dla nowych danych. Użyto: pary stowarzyszone dla klonów 50Kb i dla BAC’ów oraz markery STS. Powstało w ten sposób 3845 składowych (components) obejmujących ~2.92Gb.")
59
Asemblacja CSA, c.d. (WGA+Shredder): Dla każdej ze składowych zastosowano WGA, po poszatkowaniu danych na kawałki. Dzięki poszatkowaniu możliwa była dodatkowa korekta błędów oraz eliminacja fragmentów chimerycznych z danych HGP.
: Dla każdej ze składowych zastosowano WGA, po poszatkowaniu danych na kawałki. Dzięki poszatkowaniu możliwa była dodatkowa korekta błędów oraz eliminacja fragmentów chimerycznych z danych HGP.")
61
Ostatni krok: Mapowanie rusztowań do genomu
Do dalszej obróbki wybrano dane otrzymane z CSA. Wykorzystano dwie mapy fizyczne genomu: mapa markerów STS oraz mapa linii papilarnych BAC’ów. W ten sposób większość rusztowań została przyporządkowna pozycjom w genomie (~98% genomu). Powstało ~21,600 przerw pomiędzy rusztowaniami.
. Powstało ~21,600 przerw pomiędzy rusztowaniami.")
62
Analiza genomu (wg. Celery)
Poszukiwanie genów. Wstępny opis chromosomów. Korelacja gęstości genów z innymi wielkościami. Rozkład genów wg. molekularnej funkcji. Duplikacje genomu w skali makro.

63
Poszukiwanie genów System ekspercki Otto - symulacja czynności wykonywanych przez człowieka opisującego chromosomy. Otto wykrył 6538 genów homologicznych do znanych genów oraz 11,226 nowych fragmentów podejrzanych o bycie genem. Łącznie: 17,764 geny.

64
Poszukiwanie genów, c.d. Oprócz Otto użyto trzech programów odgadujących geny: GRAIL, Genescan, FgenesH. Zrobiły one łącznie 76,410 różnych predykcji, z czego 57,935 predykcji nie pokrywało się z predykcjami Otto. Dodatkowy filtr: co najmniej jedno potwierdzenie z następującej listy.

65
Cztery typy potwierdzeń dla predykcji genów
Homologia ze znanym białkiem. Zawieranie ludzkiego EST. Zawieranie EST gryzonia. Występowanie w genomie myszy.

66
Ile jest genów? Biorąc wszystkie predykcje Otto oraz predykcje w/w trzech programów spełniające dodatkowo warunek: Co najmniej 1 potwierdzenie: 39,114 genów Co najmniej 2 potwierdzenia: 26,383 geny. Co najmniej 3 potwierdzenia: ~23,000 genów.

67
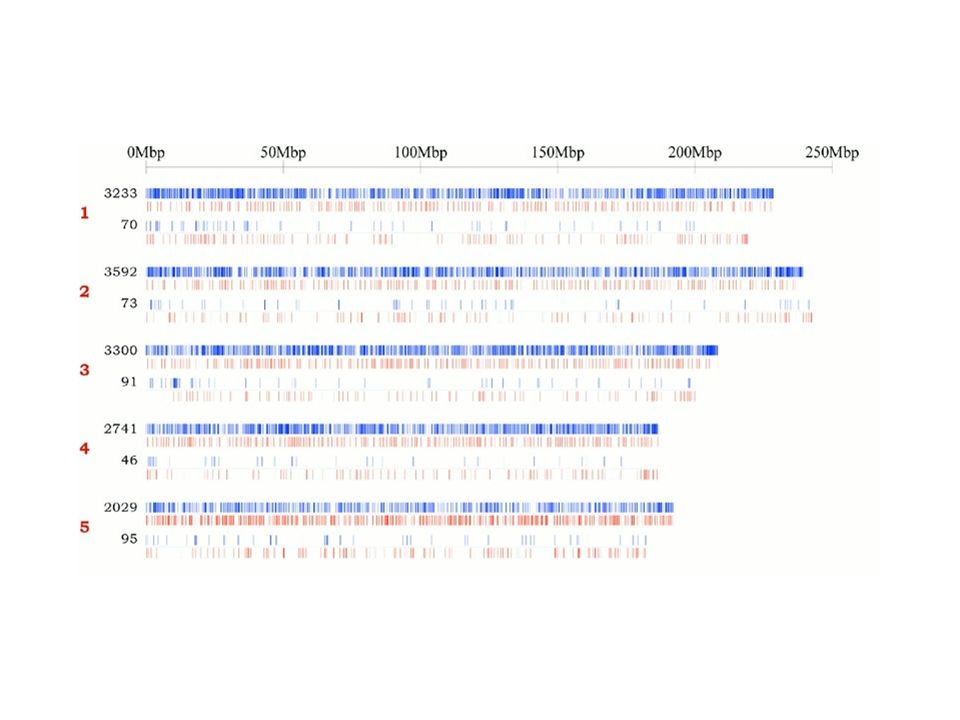
Wstępny opis Celery chromosomów
Chr. X Chr. Y

68
Chromosomy 11, 12, 13: Korelacja gęstości genów Z innymi wielkościami

69
Rozkład 26,383 genów wg. molekularnej funkcji

70
Duplikacje względem chromosomu 1

71
Duplikacje względem chromosomu 6

72
Duplikacje względem chromosomu 19 – rekordowo dużo

73
Duplikacje względem chromosomu 22 – rekordowo mało

74
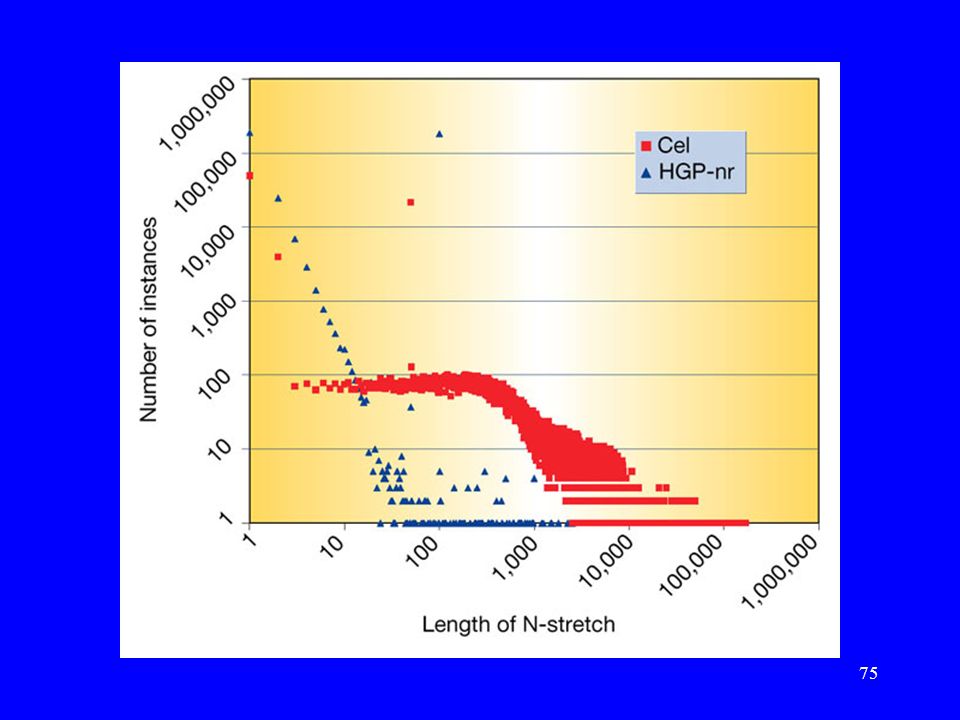
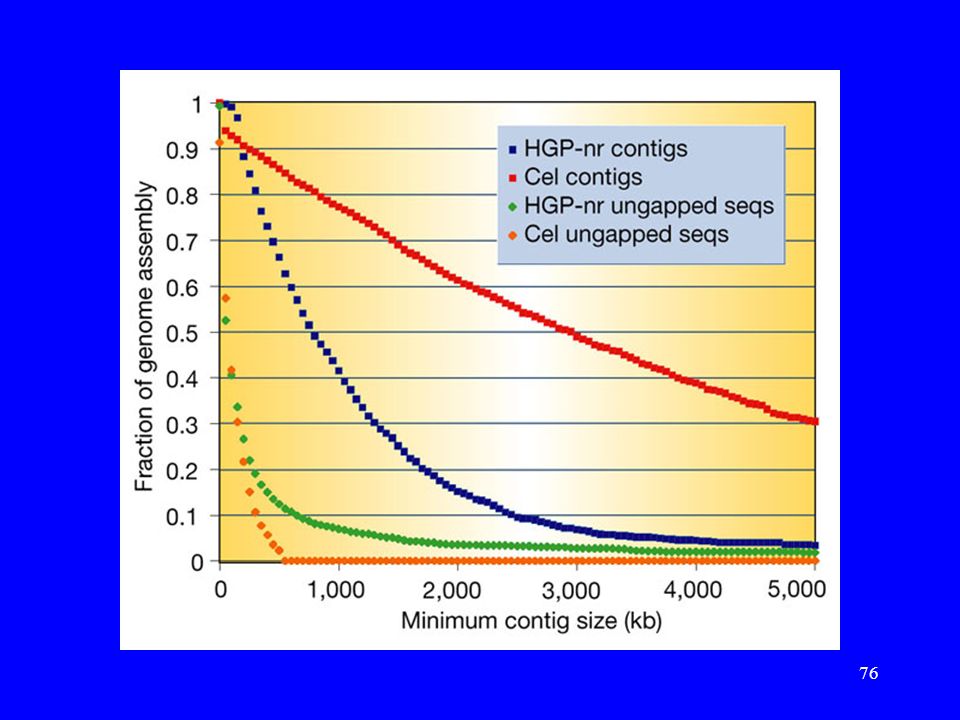
Porównanie sekwencji HGP i Celery
Praca: J. Aach, et.al. „Computational comparison of two draft sequences of the human genome.”, Nature, 409, , ( ). HGP-nr (2.9Gb). Cel Celera Genomics (Human Genome D, 2.9Gb).
. HGP-nr (2.9Gb). Cel Celera Genomics (Human Genome D, 2.9Gb).")
77
Porównania wykonane przez Celerę
Zielony kolor: sekwencje Celery są w tej samej orientacji i kolejności w obu sekwencjach. Żółty kolor: sekwencje Celery są w tej samej orientacji, ale nie w tej samej kolejności w obu sekwencjach. Czerwony kolor: sekwencje Celery nie są w tej samej orientacji w obu sekwencjach.

78
Porównania wykonane przez Celerę, c.d.
Górna część wykresu – Konsorcjum (2K, 10K, 50K). Dolna – Celera (2K, 10K, 50K). Seledynowe kreski – przerwa co najmniej b. Stowarzyszone pary (niezgodności): Czerwony – zła orientacja. Żółty – zła odległość pomiędzy końcami. Niebieskie kreski – złamania (breakpoint)
. Dolna – Celera (2K, 10K, 50K). Seledynowe kreski – przerwa co najmniej b. Stowarzyszone pary (niezgodności): Czerwony – zła orientacja. Żółty – zła odległość pomiędzy końcami. Niebieskie kreski – złamania (breakpoint)")
79
Porównanie dla chromosomu 21

80
Porównanie dla chromosomu 22

81
Porównanie dla chromosomu 19

82
Porównanie dla chromosomu 8

83
Przerwy i złamania w obu sekwencjach
Górna cześć – Konsorcjum. Dolna część – Celera. Czerwona kreska – przerwa co najmniej 10Kb. Niebieska kreska – złamanie (breakpoint): sprzeczność z co najmniej 5 stowarzyszonymi parami.
: sprzeczność z co najmniej 5 stowarzyszonymi parami.")
Podobne prezentacje













>")





