Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Wykład specjalizacyjny
BADANIE MECHANIZMÓW REAKCJI Prof. dr hab. Marianna Kańska

5
Mechanizm opisuje przebieg reakcji chemicznej.
Mechanizm reakcji Mechanizm opisuje przebieg reakcji chemicznej. Mówi on o tym: a) które wiązania ulegają pęknięciu, b) jakie wiązania się się tworzą, c) jaka jest kolejność tych zjawisk, d) z ilu etapów składa się rozpatrywany proces, e) jakie są względne szybkości poszczególnych etapów Poznanie odpowiedzi na te pytania jest często bardzo trudnym zadaniem. Szczególnie może to być skomplikowane w przypadku reakcji enzymaty-cznych, ze względu na złożoną strukturę enzymu i zachodzące procesy katalityczne.
 które wiązania ulegają pęknięciu, b) jakie wiązania się się tworzą, c) jaka jest kolejność tych zjawisk, d) z ilu etapów składa się rozpatrywany proces, e) jakie są względne szybkości poszczególnych etapów. Poznanie odpowiedzi na te pytania jest często bardzo trudnym zadaniem. Szczególnie może to być skomplikowane w przypadku reakcji enzymaty-cznych, ze względu na złożoną strukturę enzymu i zachodzące procesy katalityczne.")
6
Metody wyznaczania mechanizmów reakcji
1. Badanie produktów produktów reakcji: identyfikacja, dowody stereochemiczne. 2. Badanie produktów pośrednich: izolacja produktów pośrednich, wykrywanie produktów pośrednich (metody spektroskopowe i rezonansowe), wychwycenie produktów pośrednich (przy założeniu, że produkt pośredni będzie reagować z danym reagentem dając ściśle określony produkt), dodatek oczekiwanego produku pośredniego. 3. Badania kinetyczne: równanie kinetyczne (mechanizm musi objaśniać obserwowane równanie i rząd reakcji), badania katalizy (również inhibicji), efekty izotopowe. 4. Stosowanie cząsteczek znakowanych izotopowo (analiza produktów takimi technikami, jak MS, NMR itp.).
, wychwycenie produktów pośrednich (przy założeniu, że produkt pośredni będzie reagować. z danym reagentem dając ściśle określony produkt), dodatek oczekiwanego produku pośredniego. 3. Badania kinetyczne: równanie kinetyczne (mechanizm musi objaśniać obserwowane równanie i rząd reakcji), badania katalizy (również inhibicji), efekty izotopowe. 4. Stosowanie cząsteczek znakowanych izotopowo (analiza produktów takimi. technikami, jak MS, NMR itp.).")
7
Kinetyczny efekt izotopowy
A B A1B k2 A B A2B k1 -stała szybkości reakcji z udziałem izotopu lżejszego (1B) k2 -stała szybkości reakcji z udziałem izotopu lżejszego (2B)
 k2 -stała szybkości reakcji z udziałem izotopu lżejszego (2B)")
8
Kinetyczny efekt izotopowy kiz. lżejszego / kiz. cięższego
Jednym z najpotężniejszych narzędzi w badaniu mechanizmów reakcji jest metoda kinetycznego efektu izotopowego. Jest ona bardzo często stosowana do badania mechanizmów reakcji enzymatycznych. Teoretyczne wyjaśnienie kinetycznych efektów izotopowych jest złożonym i trudnym zadaniem. Z praktycznego punktu widzenia istotne jest jedynie uzmysłowienie sensu fizycznego tego zjawiska. Zastąpienie atomu pierwiastka, w cząsteczce związku biorącym udział w reakcji, na jego cięższy izotop często powoduje zmianę szybkości reakcji. Różna szybkość tych dwóch reakcji jest nazywana kinetycznym efektem izotopowym i określa się ją jako stosunek stałych szybkości: kiz. lżejszego / kiz. cięższego

9
Różnica ta wynika z tego, że energia oscylacyjna wiązania chemicznego na najniższym możliwym poziomie (zero-point energy) nie jest zerowa (wynosi: E = hυ) i zależy od masy zredukowanej: = zgodnie z prawem Hook’a: (k- stała siłowa niezależna od masy). Z tego wynika że wiązanie z cięższym izotopem będzie miało niższą energię oscylacji i rozerwanie wiązania będzie wymagało większej energii.
. Z tego wynika że wiązanie z cięższym izotopem będzie miało niższą energię oscylacji i rozerwanie wiązania będzie wymagało większej energii.")
10
Obrazowo przedstawiono to na poniższym schemacie:
Energia dysocjacji wiązań C-H i C-D Ta właśnie różnica w energiach dysocjacji jest powodem różnych szybkości procesów z udziałem izotopów. Efekt izotopowy obserwujemy jedynie wówczas, gdy rozpatrywany etap jest wystarczająco wolny, aby mieć decydujący wpływ na szybkość całego procesu.

11
Kinetyczne efekty izotopowe. Najważniejsze kryteria podziału.
1. Podział KEI ze względu na wielkość stosunku efekty normalne, występują wówczas gdy szybkość reakcji dla związku z izotopem lżejszym jest większa niż dla związku z izotopem cięższym brak efektu izotopowego efekty odwrotne (obserwowane rzadko) gdy:
 gdy:")
12
Metody wyznaczania mechanizmów reakcji
1. Badanie produktów produktów reakcji: identyfikacja, dowody stereochemiczne. 2. Badanie produktów pośrednich: izolacja produktów pośrednich, wykrywanie produktów pośrednich (metody spektroskopowe i rezonansowe), wychwycenie produktów pośrednich (przy założeniu, że produkt pośredni będzie reagować z danym reagentem dając ściśle określony produkt), dodatek oczekiwanego produku pośredniego. 3. Badania kinetyczne: równanie kinetyczne (mechanizm musi objaśniać obserwowane równanie i rząd reakcji), badania katalizy (również inhibicji), efekty izotopowe. 4. Stosowanie cząsteczek znakowanych izotopowo (analiza produktów takimi technikami, jak MS, NMR itp.).
, wychwycenie produktów pośrednich (przy założeniu, że produkt pośredni będzie reagować. z danym reagentem dając ściśle określony produkt), dodatek oczekiwanego produku pośredniego. 3. Badania kinetyczne: równanie kinetyczne (mechanizm musi objaśniać obserwowane równanie i rząd reakcji), badania katalizy (również inhibicji), efekty izotopowe. 4. Stosowanie cząsteczek znakowanych izotopowo (analiza produktów takimi. technikami, jak MS, NMR itp.).")
13
3. Podział KEI ze względu na położenie znacznika izotopowego w stosunku do
miejsca w cząsteczce, gdzie zachodzi etap determinujący szybkość reakcji: a) pierwszorzędowe, b) a-drugorzędowe c) b-drugorzędowe Na przykładzie mechanizmu E1 można wyjaśnić te efekty. Według tego mechanizmu, najwolniejszym etapem jest rozerwanie wiązania pomiędzy atomem węgla a grupą X (odchodzącą), co prowadzi do utworzenia karbokationu. Ten etap będzie decydował o szybkości całego procesu. W związku z tym podstawienie atomu 12C1 izotopem 14C spowolni reakcję. Taki efekt izotopowy jest nazywany efektem pierwszo-rzędowym. Analogicznie dla wodoru H2 wystąpi efekt a-drugorzędowy, a dla wodoru H3 wystąpi efekt b-drugorzędowy. Mechanizm E1
 pierwszorzędowe, b) a-drugorzędowe. c) b-drugorzędowe. Na przykładzie mechanizmu E1 można wyjaśnić te efekty. Według tego mechanizmu, najwolniejszym etapem jest rozerwanie wiązania pomiędzy atomem węgla a grupą X (odchodzącą), co prowadzi do utworzenia karbokationu. Ten etap będzie decydował o szybkości całego procesu. W związku z tym podstawienie atomu 12C1 izotopem 14C spowolni reakcję. Taki efekt izotopowy jest nazywany efektem pierwszo-rzędowym. Analogicznie dla wodoru H2 wystąpi efekt a-drugorzędowy, a dla wodoru H3 wystąpi efekt b-drugorzędowy. Mechanizm E1.")
14
4. Podział na efekty substratowe i rozpuszczalnikowe
substratowe – występują wówczas gdy zmiana składu izotopowego substratu powoduje zmianę szybkości reakcji, rozpuszczalnikowe – występuje wówczas gdy zmiana rozpuszczalnika np. z H2O na D2O powoduje zmianę szybkości reakcji. 5. Podział efektów izotopowych znajdujących odbicie w zmianie kinetycznych parametrów reakcji enzymatycznych: kinetyczne efekty izotopowe na Vmax kinetyczne efekty izotopowe na Vmax /Km

15
4. Podział na efekty substratowe i rozpuszczalnikowe
substratowe – występują wówczas gdy zmiana składu izotopowego substratu powoduje zmianę szybkości reakcji, rozpuszczalnikowe – występuje wówczas gdy zmiana rozpuszczalnika np. z H2O na D2O powoduje zmianę szybkości reakcji. 5. Podział efektów izotopowych znajdujących odbicie w zmianie kinetycznych parametrów reakcji enzymatycznych: kinetyczne efekty izotopowe na Vmax kinetyczne efekty izotopowe na Vmax /Km

16
METODY WYZNACZANIA KINETYCZNYCH EFEKTÓW IZOTOPOWYCH
1. Bezpośrednie wyznaczanie kinetycznych efektów izotopowych. 2. Metoda zaburzeń równowagi. 3. Metody z użyciem spektrometrii mas.

17
Wyznaczanie KIE wg równań Bigeleisena i Wolsgerga
- R0 - aktywność molową lub stosunek zawartości izotopu lżejszego do izotopu cięższego w substracie przed rozpoczęciem reakcji, - Rp - aktywność molową lub stosunek zawartości izotopu lżejszego do izotopu cięższego w produkcie w chwili, gdy stopień przereagowania wynosi f, - Rs - aktywność molową lub stosunek zawartości izotopu lżejszego do izotopu cięższego w substracie, gdy stopień przereagowania wynosi f, - f - stopień przereagowania. - α - kinetyczny efekt izotopowy,

22
Mechanizm kondensacji Dieckmana

24
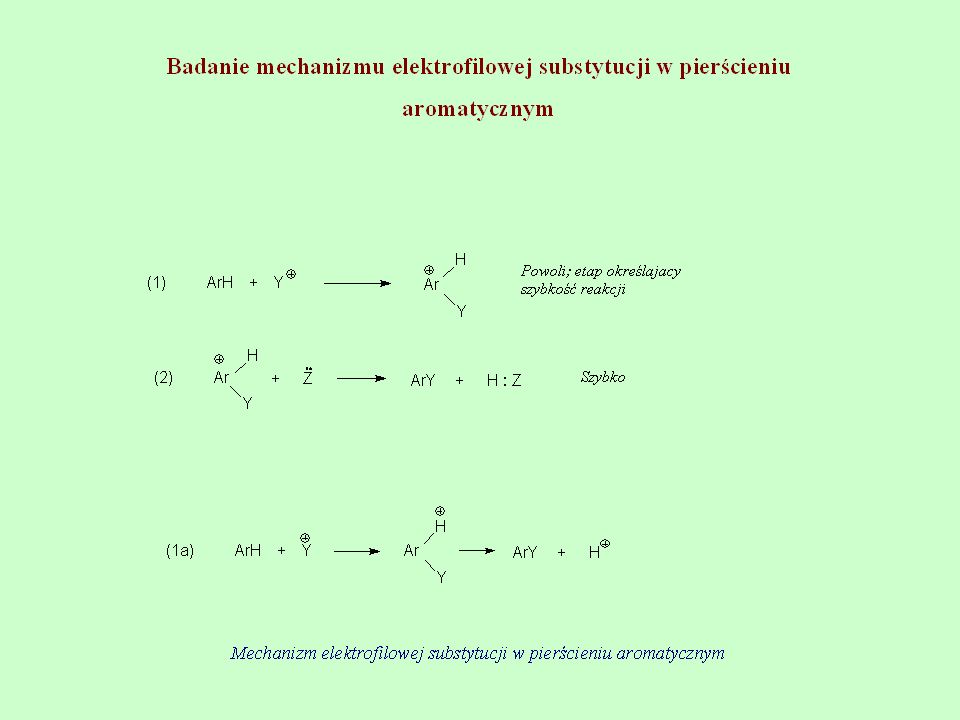
Badanie mechanizmu reakcji addycji elektrofilowej chlorku
2,4 dinitrobenzenosulfenowego do styrenu i jego para pochodnych w środowisku kwasu octowego Mechanizm reakcji addycji elektrofilowej chlorku 2,4-dinitrobenzenosulfenowego

25
Jeżeli reakcje addycji chlorku 2,4-dinitrobenzenosulfenylowego do styrenu i jego para pochodnych prowadzi się w kwasie octowym, to wiadomo, że reakcja przebiega zgodnie z regułą Markownikowa i dodatnia część cząsteczki chlorku 2,4-dinitrobenzenosulfenowego przyłącza się do βC natomiast ujemny chlor przyłącza się do αC i powstają odpowiednie siarczki chloro fenyloetylowo-2,4-dinitrofenylowe. Powstaje pytanie, jaką strukturę posiada kompleks aktywny powstający w etapie określającym szybkość reakcji w reakcji elektrofilowej? Prezentowany schemat zawiera trzy różne struktury stanów przejściowych dające ten sam produkt końcowy. Na temat reakcji elektrofilowej addycji do nienasyconych węglowodorów ukazało się wiele prac, ale nie było jednomyślności jaką strukturę ma kompleks aktywny. Problem ten mógł być rozwiązany przez wyznaczenie KEI 14C w pozycji α- i β-styrenów zawierających elektronodonorowe i elektronoakceptorowe podstaw\niki. Przewidziano, że jeżeli kompleks aktywny posiada strukturę (2) to powinniśmy obserwować kinetyczny efekt izotopowy dla βC, ponieważ tworzy się wiązanie z siarką tylko przy tym węglu. Natomiast jeżeli kompleks aktywny posiada strukturę (3) bądź (4) wówczas powinniśmy obserwować KEI dla αC i dla βC.
 to powinniśmy obserwować kinetyczny efekt izotopowy dla βC, ponieważ tworzy się wiązanie z siarką tylko przy tym węglu. Natomiast jeżeli kompleks aktywny posiada strukturę (3) bądź (4) wówczas powinniśmy obserwować KEI dla αC i dla βC.")
26
Badania doprowadziły do wyznaczenia KEI dla αC i βC następujących dla kolejno podstawionych styrenów: αC p-CH3; p-H; p-Cl; k/kα = 1,004; 1,022; 1,027 βC p-CH3; p-H: p-Cl; k/kβ = 1,037; 1,032; 1,035 Oznaczenia wartości k/kα i k/kβ wykazały, że kinetyczny efekt izotopowy dla węgla 14C jest zależny od miejsca podstawienia izotopowego oraz od charakteru podstawników w pierścieniu aromatycznym. Wyznaczona wartość k/kβ dla βC jest dość duża i nie zależny od charakteru podstawników w pozycji para pierścienia. Natomiast k/kα jest zależny od charakteru podstawnika. Wyraźnie mały kinetyczny efekt izotopowy węgla 14C w reakcji addycji ArSCl do styrenu, posiadający elektronodonorowy podstawnik w pozycji para pierścienia aromatycznego, sugeruje, że struktura stanu przejściowego jest zbliżona do struktury otwartej karbokationu (2), w której dodatni ładunek jest zlokalizowany przy węglu α. Wiązanie βC-S tworzy się niezależnie od mechanizmu i dlatego jest jasne, że KEI występuje i jego wartość nie zmienia się, niezależnie od tego jaki podstawnik jest w pierścieniu aromatycznym.
, w której dodatni ładunek jest zlokalizowany przy węglu α. Wiązanie βC-S tworzy się niezależnie od mechanizmu i dlatego jest jasne, że KEI występuje i jego wartość nie zmienia się, niezależnie od tego jaki podstawnik jest w pierścieniu aromatycznym.")
27
Jeśli aktywny kompleks miałby strukturę (3) lub (4) to utworzone wiązanie pomiędzy αC i siarką powinno być taki samo lub podobne i wówczas KEI dla αC powinien być podobny. Im silniejsze jest wiązanie αC-S tym większy powinien być KEI. Jeżeli ładunek dodatni na αC jest bardziej zdelokalizowany w pierścieniu wówczas wiązanie αC-S jest bardzo słabe lub go nie ma i wtedy jest brak kinetycznego efektu izotopowego. Jeżeli podstawnik jest elektronodonorowy (-CH3), to wolna para elektronowa jest do pewnego stopnia zdelokalizowana, co powoduje zwiększenie chmury elektronowej pierścienia, a następnie osłabienie ładunku dodatniego przy αC. Wiązanie αC-S jest wtedy bardzo słabe i w konsekwencji tego KEI jest bardzo mały. Obecność chloru w pozycji para pierścienia powoduje, że gęstość elektronowa w pierścieniu jest mniejsza niż w cząsteczce styrenu i dlatego też wiązanie αC-S jest silniejsze i KEI jest większy. A więc jeżeli podstawnik jest elektronodonorowy, to aktywny kompleks ma strukturę (2). Jeżeli podstawnik jest elektronoakceptorowy, to aktywny kompleks ma strukturę (3) lub (4). Reasumując, struktura kompleksu aktywnego powstającego w etapie określającym szybkość reakcji zależy od budowy podstawnika znajdującego się przy podwójnym wiązaniu.
. Jeżeli podstawnik jest elektronoakceptorowy, to aktywny kompleks ma strukturę (3) lub (4). Reasumując, struktura kompleksu aktywnego powstającego w etapie określającym szybkość reakcji zależy od budowy podstawnika znajdującego się przy podwójnym wiązaniu.")
28
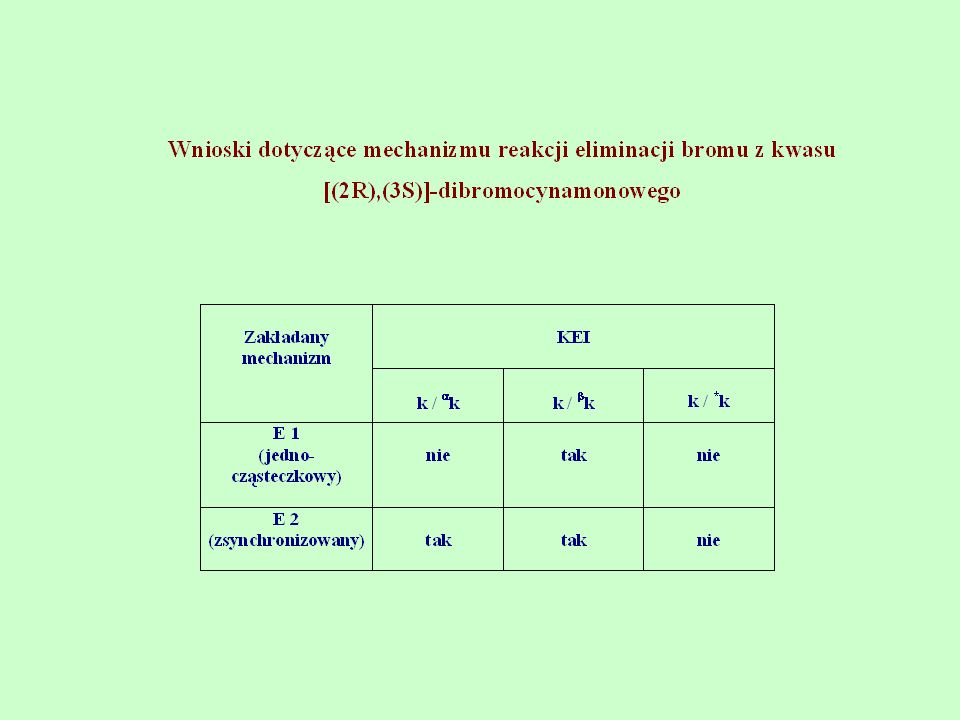
Badania mechanizmu reakcji eliminacji bromu z kwasów dibromocynamonowych do odpowiednich kwasów cynamonowych

29
Mechanizm eliminacji kwasu para metylo[(2R),(3S)]-dibromocynamonowego
![Mechanizm eliminacji kwasu para metylo[(2R),(3S)]-dibromocynamonowego](http://slideplayer.pl/slide/418468/1/images/29/Mechanizm+eliminacji+kwasu+para+metylo%5B%282R%29%2C%283S%29%5D-dibromocynamonowego.jpg "Mechanizm eliminacji kwasu para metylo[(2R),(3S)]-dibromocynamonowego")
30
Mechanizm eliminacji kwasu para metylo[(2R),(3S)]-dibromocynamonowego
Badania wykazały, że kinetyczny efekt izotopowy 14C występuje w pozycjach α, β, oraz jest zależny od miejsca podstawienia izotopowego i od charakteru podstawnika w pierścieniu aromatycznym. Gdy: R = H, p-CH3, oraz p-NO2 wtedy (k12\k14) w pozycji wynoszą odpowiednio: 1,05226; 1,0094; 1,0233. Natomiast (k12\k14) w pozycji dla podstawników R = p-CH3 i H wynoszą odpowiednio: 1,072; 1,0483
![Mechanizm eliminacji kwasu para metylo[(2R),(3S)]-dibromocynamonowego](http://slideplayer.pl/slide/418468/1/images/30/Mechanizm+eliminacji+kwasu+para+metylo%5B%282R%29%2C%283S%29%5D-dibromocynamonowego.jpg "Badania wykazały, że kinetyczny efekt izotopowy 14C występuje w pozycjach α, β, oraz jest zależny od miejsca podstawienia izotopowego i od charakteru podstawnika w pierścieniu aromatycznym. Gdy: R = H, p-CH3, oraz p-NO2. wtedy (k12\k14) w pozycji wynoszą odpowiednio: 1,05226; 1,0094; 1,0233. Natomiast (k12\k14) w pozycji dla podstawników R = p-CH3 i H wynoszą odpowiednio: 1,072; 1,0483.")
32
Badanie mechanizmu eliminacji amin z soli p-nitrofenylo-2-etylo-N,N,N-trimetyloamoniowej i n-propylo-N,N,N-trimetyloamoniowej Reakcje eliminacji, badane metodą KEI z zastosowaniem ciężkich atomów zachodziły głownie według mechanizmu E1 i E2. W związku z tym prowadzone badania były głównie ukierunkowane w stronę wyznaczenia trwałości wiązań przy βC-H i αC-X. Skomplikowana natura takiej reakcji została wyjaśniona na przykładzie wyznaczenia KEI dla kolejno znakowanych związków w trakcie rozkładu soli n-propylo-N,N,N-trimetyloamoniowej oraz p-nitrofenylo-2-etylo-N,N,N-trimetylo-amoniowej Mechanizm eliminacji soli amoniowych do styrenu

33
i kH/kT = 2,12 dla trytu w pozycji β.
Badano kinetyczny efekt izotopowy dla węgla 14C, wodoru i azotu. W literaturze występują znaczne różnice w wyznaczonych efektach izotopowych przez dwie oddzielne grupy badawcze. Pierwsza grupa dla podstawnika R = CH3 otrzymała: k/kβ = 1,036 dla 14C w pozycji β, k/kα = 1,069 dla 14C w pozycji α, oraz kH/kT = dla trytu w pozycji β. Reakcja ta była prowadzona w temperaturze 50 oC. Ponadto wyznaczono KEI dla reakcji w tych samych warunkach z podstawnikiem R = p-NO2C6H4 dla 14C w pozycji α, gdzie otrzymano: k/kα = 1,026. Z tego widać, że występujące znaczne efekty izotopowe przy αC, βC i βH wpływają na etapy determinujące szybkość reakcji. Druga grupa badawcza dla podstawnika R = p-NO2C6H4 wyznaczyła kinetyczny efekt izotopowy: k/kα = 1, dla14C w pozycji 2, k14/k15 = 1, dla azotu, i kH/kT = 2,12 dla trytu w pozycji β. Reakcję prowadzono w temperaturze 100 oC. Przyczyna tych rozbieżności nie jest znana, ale autorzy wyciągają podobne wnioski, że zmiany wiązań przy N, αC,βC i βH decydują o szybkości reakcji.

34
Badanie mechanizmu reakcji dehydrohalogenacjii

35
Badanie mechanizmu reakcji dehydrohalogenacjii
Zaproponowany mechanizm reakcji dehydrohalogenacji przedstawia poniższy schemat Przed dokładnym przebadaniem reakcji dehydrohalogenacji sądzono, że przebiega ona w środowisku zasadowym według mechanizmu E1cB, ale nie wykluczono również mechanizmu podobnego do E2. W związku z czym przebadano proces eliminacji z użyciem czterech uprzednio podanych układów.

36
Zakładany mechanizm eliminacji amoniaku i odtworzenie miejsca aktywnego

37
Mechanizm reakcji eliminacji z udziałem PAL zaproponowany przez Havir’a i Hanson’a

38
Mechanizm reakcji eliminacji z udziałem PAL zaproponowany przez Schuster’a i Retey’a

39
Kinetyczny efekt izotopowy H/T w pozycji 3-pro-S L-tyrozyny
Liaza fenyloalaninowa katalizuje również eliminację amoniaku z L-tyrozyny, co pozwala na zbadanie wpływu grupy elektrodonorowej na wielkość kinetycznego efektu izotopowego w tej reakcji. Nie można jednocześnie wykluczyć, że reakcja eliminacji z udziałem L-tyrozyny przebiega według innego mechanizmu. Potwierdzeniem takiej tezy byłby wynik znacząco różny od otrzymanego dla L-Phe, czyli na przykład brak efektu lub duży efekt.

40
Kinetyczny efekt izotopowy H/T w pozycji orto pierścienia aromatycznego L-fenyloalaniny
Wyniki badań kinetycznego efektu izotopowego H/T w pozycji 2 i 6 pierścienia aromatycznego L-fenyloalaniny.

41
Kinetyczny efekt izotopowy 12C/14C w pozycji 2 L-Phe
Kinetyczny efekt izotopowy 12C/14C w pozycji 2 L-fenyloalaniny

42
Procedura wyznaczenie KEI H/T w pozycji 3-pro-R

43
Kinetyczny efekt izotopowy H/T w pozycji 3-pro-R L-Phe

44
Procedura badania KEI w pozycji 3-pro-S L-Phe

45
Kinetyczny efekt izotopowy D/T w pozycji 3-pro-S L-Phe

46
Zależność Swain’a-Schaad’a
α = α = Gdzie: kH/kT - KIE dla 1H/3H. kH/kD - KIE dla 1H/2H. kD/kT - KIE dla 2H/3H. Jeśli efekt 1H/3H, obliczony z efektów 1H/2H lub 2H/3H przy pomocy wspomnianych zależności, jest mniejszy od efektu zaobserwowanego, wtedy prawdopodobnie w reakcji następuje tunelowanie protonu. Jeśli wartość wyliczonego KIE jest większa od zaobserwowanej, to mamy do czynienia ze złożonością kinetyczną, tzn. nie tylko etap odrywania protonu decyduje o szybkości reakcji.

Podobne prezentacje