Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
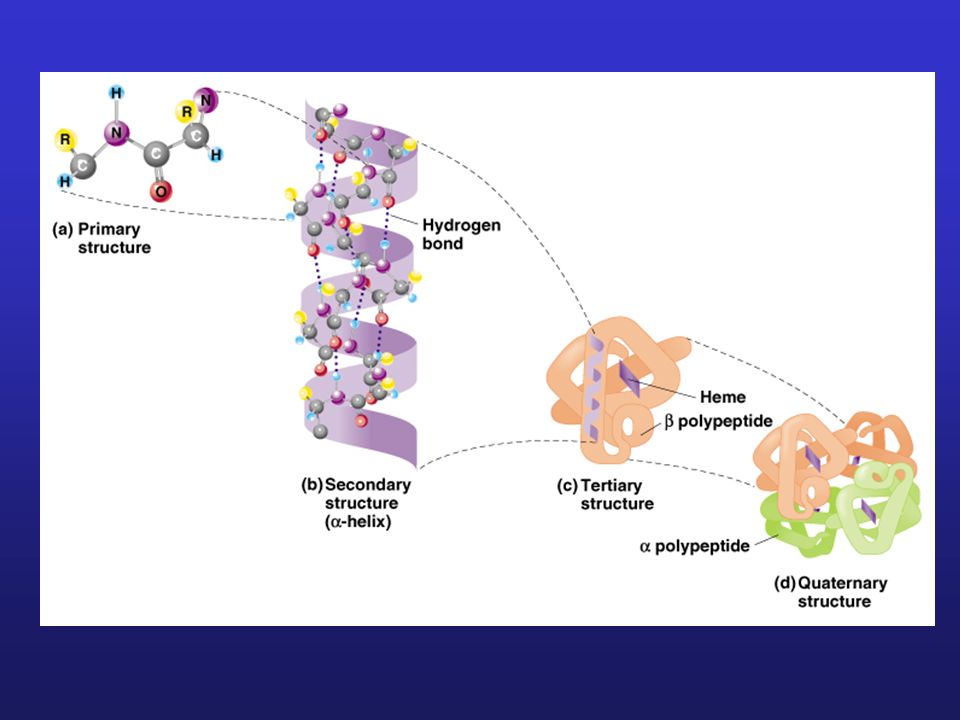
Metody określania struktury enzymów
Enzymologia-3 Metody określania struktury enzymów

2
Metody badania struktury przestrzennej (konformacji) enzymów można podzielić na dwie zasadnicze grupy: metody bezpośrednie (rentgenografia strukturalna, NMR) pośrednie (empiryczne, modyfikacje chemiczne, ukierunkowana mutageneza, metody spektroskopowe).
 pośrednie (empiryczne, modyfikacje chemiczne, ukierunkowana mutageneza, metody spektroskopowe).")
4
1. Określanie struktury I- i II-rzędowej
1.1 Określanie składu aminokwasowego i masy cząsteczkowej białka 1.2 Zastosowanie spektrometrii mas w analizie białek 1.3 Sekwencjonowanie białek 1.4 Identyfikacja wiązań disiarczkowych 1.5 Spektroskopowe metody określania struktury II-rzędowej

5
Określanie składu aminokwasowego
Hydroliza białka do składowych aminokwasów Warunki klasyczne: 6 M HCl zawierający 0.1% phenol, w próżni lub atmosferze azotu, w zamkniętej ampułce, 110 C, 18 – 96 h Problemy i ograniczenia: częściowa destrukcja reszt Ser, Thr, Tyr. Zakłada się 10% destrukcję Ser oraz 5% destrukcję Thr i Tyr w ciągu 24 h, lub przeprowadza się hydrolizę trzech próbek, odpowiednio przez 24, 48 i 72 h i następnie ekstrapoluje się wyniki dla momentu zero reszty Asn i Gln są przekształcane w Asp i Glu. Białko poddaje się działaniu bis (1,1-trifluoro-acetoksy)jodobenzenu /36 mg/ml in 0.01 M TFA, 4 h, 60 C, w ciemności/. W tych warunkach reszty karboksyamidowe są przekształcane w aminy całkowita destrukcja Trp Konieczne przeprowadzenie osobnej hydrolizy. Warunki: 3 M kwas p-toluenosulfonowy, 110 C, 24 – 72 h, w prózni
jodobenzenu /36 mg/ml in 0.01 M TFA, 4 h, 60 C, w ciemności/. W tych warunkach reszty karboksyamidowe są przekształcane w aminy. całkowita destrukcja Trp. Konieczne przeprowadzenie osobnej hydrolizy. Warunki: 3 M kwas p-toluenosulfonowy, 110 C, 24 – 72 h, w prózni.")
6
Separacja i analiza mieszanin aminokwasów
Separacja mieszaniny PTC-aminokwasów metodą HPLC. Wypełnienie: octadecylosilan, elucja mieszaniną acetonitryl /woda Chromatografia jonowymienna mieszaniny aminokwasów. Kolumna Dowex 50WX4 Derywatyzacja aminokwasów Analizatory jonowymienne – po kolumnie Ninhydryna; barwne pochodne, detekcja przy 570 nm lub 440 nm (Pro); fluoreskamina lub aldehyd ftalowy + 2-tioetanol; produkty fluoryzujące; HPLC w układzie faz odwróconych – przed kolumną fenylotioizocyjanian; PTC-pochodne chlorek dansylu; DNS-pochodne chlorek N-(9-fluorenylometoksykarbonylu; FMOC-pochodne
; fluoreskamina lub aldehyd ftalowy + 2-tioetanol; produkty. fluoryzujące; HPLC w układzie faz odwróconych – przed kolumną. fenylotioizocyjanian; PTC-pochodne. chlorek dansylu; DNS-pochodne. chlorek N-(9-fluorenylometoksykarbonylu; FMOC-pochodne.")
7
Metody określania masy cząsteczkowej białek
Metody bezpośrednie: sedymentacja (wirowanie strefowe) spektrometria mas (MS ESI) Metody porównawcze: elektroforeza w żelu poliakrylamidowym (SDS-PAGE i natywna) chromatografia rozmiarów wykluczających
 spektrometria mas (MS ESI) Metody porównawcze: elektroforeza w żelu poliakrylamidowym (SDS-PAGE i natywna) chromatografia rozmiarów wykluczających.")
8
wzorcowa krzywa kalibracyjna log MW = f (Ve/V0).
V0 oznacza objętość swobodną kolumny Veobjętość elucji anhydraza węglanowa (29 kDa), BSA (66 kDa), dehydrogenaza alkoholowa (150 kDa), - amylaza (200 kDa), apofferrytyna (443 kDa), tyroglobulina (669 kDa).
, BSA (66 kDa), dehydrogenaza alkoholowa (150 kDa), - amylaza (200 kDa), apofferrytyna (443 kDa), tyroglobulina (669 kDa).")
9
Schemat blokowy spektrometru mas

10
Metody jonizacji Elektrorozpylanie (Electrospray, ESI), polegające na rozpylaniu cieczy zawierającej badaną substancję z igły, do której przyłożono wysokie napięcie (zwykle kV) pod ciśnieniem atmosferycznym. Jonizacja elektronami (Electron Ionisation - EI) - jonizacja przy pomocy wiązki elektronów. Jonizacja odbywa się w próżni. Metoda ta powoduje zwykle fragmentację badanych cząsteczek. EI charakteryzuje się stosunkowo małą wydajnością - poniżej 1% cząsteczek ulega jonizacji.
, polegające na rozpylaniu cieczy zawierającej badaną substancję z igły, do której przyłożono wysokie napięcie (zwykle kV) pod ciśnieniem atmosferycznym. Jonizacja elektronami (Electron Ionisation - EI) - jonizacja przy pomocy wiązki elektronów. Jonizacja odbywa się w próżni. Metoda ta powoduje zwykle fragmentację badanych cząsteczek. EI charakteryzuje się stosunkowo małą wydajnością - poniżej 1% cząsteczek ulega jonizacji.")
11
Metody jonizacji Bombardowanie szybkimi atomami (Fast-Atom Bombardment FAB), polegającą na bombardowaniu cząsteczki obojętnymi atomami o wysokiej energii (zwykle 17 lub 70 eV). Cząsteczki mogą znajdować się w fazie gazowej lub być rozpuszczone w ciekłej, mało lotnej substancji (matrycy) np. glicerolu. Desorpcja laserowa z udziałem matrycy (Matrix Assisted Laser Desorption Ionisation - MALDI) - w której stosuje się jonizację laserową, ale z tak dobraną energią wiązki, aby nie doprowadzać do fragmentacji cząsteczek (łagodna metoda jonizacji), lecz tylko do ich "wybijania" ze specjalnie przygotowanej matrycy. Matryca absorbuje energię lasera, która jest później przekazywana do analizowanych cząsteczek.
, polegającą na bombardowaniu cząsteczki obojętnymi atomami o wysokiej energii (zwykle 17 lub 70 eV). Cząsteczki mogą znajdować się w fazie gazowej lub być rozpuszczone w ciekłej, mało lotnej substancji (matrycy) np. glicerolu. Desorpcja laserowa z udziałem matrycy (Matrix Assisted Laser Desorption Ionisation - MALDI) - w której stosuje się jonizację laserową, ale z tak dobraną energią wiązki, aby nie doprowadzać do fragmentacji cząsteczek (łagodna metoda jonizacji), lecz tylko do ich wybijania ze specjalnie przygotowanej matrycy. Matryca absorbuje energię lasera, która jest później przekazywana do analizowanych cząsteczek.")
12
Analizator masy Analizator czasu przelotu (Time Of Flight TOF) - jony wprowadzane do analizatora są przyspieszane przy pomocy impulsu elektrycznego i zaczynają dryfować przez komorę analizatora. Na końcu analizatora znajduje się detektor jonów połączony z urządzeniem rejestrującym czas od impulsu przyspieszającego do momentu uderzenia określonego jonu w detektor. Pomiar m/z jest oparty na fakcie, że ze wzrostem masy cząsteczkowej jonów, wydłuża się ich czas przelotu.
 - jony wprowadzane do analizatora są przyspieszane przy pomocy impulsu elektrycznego i zaczynają dryfować przez komorę analizatora. Na końcu analizatora znajduje się detektor jonów połączony z urządzeniem rejestrującym czas od impulsu przyspieszającego do momentu uderzenia określonego jonu w detektor. Pomiar m/z jest oparty na fakcie, że ze wzrostem masy cząsteczkowej jonów, wydłuża się ich czas przelotu.")
13
Schemat spektrometru MALDI-TOF

14
Sekwencjonowanie białek
Rozszczepienie łańcucha białka na fragmenty metodami enzymatycznymi lub chemicznymi Separacja i izolacja peptydów Sekwencjonowanie peptydów Określenie kompletnej sekwencji łańcuch białka na podstawie porównania nakładających się sekwencji peptydów

15
Enzymatyczne metody rozszczepienia białek
Zasady ogólne: białka należy zdenaturować przed poddaniem ich trawieniu enzymatycznemu należy używać lotnych buforów (można je usunąć poprze liofilizację) należy używać enzymów proteolitycznych specjalnej jakości (sequencing grade) warunki reakcji powinny być zoptymalizowane Enzymy wykorzystywane do specyficznego cięcia: Trypsyna – karboksylowa strona reszt Arg, Lys i S-aminoetylocysteiny; specyficzność można ograniczyć do Arg poprzez modyfikacje reszt lizyny np, bezwodnikiem kwasu bursztynowego; Chymotrypsyna – karboksylowa strona Phe, Trp, Tyr and Leu (w kolejności zmniejszającej się podatności) Termolizyna – aminowa strona Leu, Ile, Phe, Val Proteaza V8 ze S. aureus – wiązania Glu-X oraz (znacznie mniej efektywnie) Asp-X Endoproteinaza Lys-C – wiązania Lys-X
 należy używać enzymów proteolitycznych specjalnej jakości (sequencing grade) warunki reakcji powinny być zoptymalizowane. Enzymy wykorzystywane do specyficznego cięcia: Trypsyna – karboksylowa strona reszt Arg, Lys i S-aminoetylocysteiny; specyficzność. można ograniczyć do Arg poprzez modyfikacje reszt lizyny np, bezwodnikiem. kwasu bursztynowego; Chymotrypsyna – karboksylowa strona Phe, Trp, Tyr and Leu (w kolejności zmniejszającej. się podatności) Termolizyna – aminowa strona Leu, Ile, Phe, Val. Proteaza V8 ze S. aureus – wiązania Glu-X oraz (znacznie mniej efektywnie) Asp-X. Endoproteinaza Lys-C – wiązania Lys-X.")
16
Metody chemicznego cięcia łańcuchów białkowych
1. Indukowane działaniem CNBr rozszczepienie przy resztach Met

17
2. Rozszczepienie wiązań Asn-Gly działaniem N-hydroksyloaminy
Warunki : 6 M chlorowodorek guanidyny, 2 M chlorowodorek hydroksyloaminy, pH = 9.0, w obecności 4.5 M LiOH

18
Sekwencjonowanie białek
Degradacja Edmana N C S + H 2 R 1 O pH 8 F 3 bezwodny rozcieńczony kwas mineralny fenylotiohydantoina (PTH) n-peptyd (n-1)-peptyd kolejny PITC cykl (DABITC) (DNS) DNS-pochodne aminokwasów można wykryć na poziome fentomolowym Metoda DABITC/PITC DABITC tworzy barwne pochodne z aminokwasami; wysoka czułość, ale tylko 60% efektywność sprzęgania. Metoda DABITC/PITC – każdy cykl składa się z dwóch etapów sprzęgania: DABITC, a następnie PITC
 n-peptyd. (n-1)-peptyd. kolejny. PITC. cykl. (DABITC) (DNS) DNS-pochodne aminokwasów. można wykryć na poziome. fentomolowym. Metoda DABITC/PITC. DABITC tworzy barwne pochodne z aminokwasami; wysoka czułość, ale tylko 60% efektywność sprzęgania. Metoda DABITC/PITC – każdy cykl składa się z dwóch. etapów sprzęgania: DABITC, a następnie PITC.")
19
Oznaczanie N-końca

20
Oznaczanie C-końca

21
Mikrosekwencjonowanie peptydów
Możliwość analizy próbek w ilościach nanogramowych

22
Ustalenie sekwencji białka oligomerycznego
Dysocjacja poszczególnych podjednostek: Jeśli podjednostki połączone są tylko wiązaniami niekowalencyjnymi, wystarczy zastosować czynniki denaturujące, takie jak roztwór 8M mocznika lub roztwór 6M chlorowodorku guanidyny, żeby doprowadzić do ich dysocjacji. Jeśli istnieją w białku wiązania disiarczkowe, należy je zerwać na drodze redukcji białka, b-merkaptoetanol i ditiotreitol, mogą całkowicie redukować mostki disiarczkowe w białku do grup –SH

23
Zabezpieczenie wolnych Cys przed rekombinacją
Uwolnione polipeptydy rozdziela się np. chromatograficznie, po czym można realizować procedurę, zmierzającą do ustalenia ich sekwencji,

24
Zastosowanie spektrometrii mas do analizy peptydów
1. MS-FAB (Fast Atom Bombardment) Materiał analizowany: peptydy powstające w wyniku rozszczepienia chemicznego lub enzymatycznego, rozpuszczone w 30% kwasie octowym (0.5 – 1 g/l); taki roztwór miesza się z glicerolem (50:50, v/v), Próbka nie może zawierać soli Na próbkę działa się strumieniem atomów Ar lub Xe lub jonami Cs+ (5 – 10 kV); Selekcja jonów: analizator sektorów magnetycznych lub TOF 2. MS MALDI-TOF Materiał analizowany: Mieszaniny peptydów powstające w wyniku traktowania białka kilkoma różnymi enzymami proteolitycznymi (osobne próbki). Zwykle wstępnie blokuje się reszty cysteiny i/lub lizyny. Próbka w matrycy stałej. Złożone widma analizowane są w celu identyfikacji poszczególnych peptydów. Metodę można także stosować do identyfikacji miejsc modyfikacji białek
 Materiał analizowany: peptydy powstające w wyniku rozszczepienia chemicznego. lub enzymatycznego, rozpuszczone w 30% kwasie octowym (0.5 – 1 g/l); taki roztwór. miesza się z glicerolem (50:50, v/v), Próbka nie może zawierać soli. Na próbkę działa się strumieniem atomów Ar lub Xe lub jonami. Cs+ (5 – 10 kV); Selekcja jonów: analizator sektorów magnetycznych lub TOF. 2. MS MALDI-TOF. Materiał analizowany: Mieszaniny peptydów powstające w wyniku traktowania białka. kilkoma różnymi enzymami proteolitycznymi (osobne próbki). Zwykle wstępnie blokuje. się reszty cysteiny i/lub lizyny. Próbka w matrycy stałej. Złożone widma analizowane są w celu identyfikacji poszczególnych peptydów. Metodę można także stosować do identyfikacji miejsc modyfikacji białek.")
25
MS-FAB w analizie peptydów
Widmo MS FAB peptydu Arg-Pro-Val-Trp-Pro- Asn-Gly-Ala-Glu-Ser-Ala-Glu-Ala-Phe-Pro-Leu-Glu-Phe. Mniejszy rysunek – powiększenie obszaru (M + H)+ Sekwencjonowanie białek z zastosowaniem tandemowej spektrometrii MS-FAB
+ Sekwencjonowanie białek z zastosowaniem tandemowej. spektrometrii MS-FAB.")
26
Analiza MS MALDI-TOF mieszanin peptydów otrzymanych w wyniku działania chymotrypsyny
na enzym, syntazę GLcN-6-P: A – forma natywna; B – forma poddana działaniu inaktywatora lączącego się z enzymem wiązaniem kowalencyjnym Wynik nałożenia obszarów 200 – 800 widm A i B. Sygnałów obecnych w obu widmach nie pokazano

27
Identyfikacja wiązań disiarczkowych
1. Czy enzym zawiera w swojej strukturze wiązania disiarczkowe? Należy porównać ruchliwość elektroforetyczną (SDS-PAGE) enzymu poddanego uprzednio denaturacji w warunkach redukujących (w obecności merkaptoetanolu) i nieredukujących 2. Określenie liczby wiązań –S-S- Przygotować dwie próbki enzymu w roztworze zawierającym 6 M chlorowodorek guanidyny Poddać próbkę 1 działaniu 10 mM DTT; W tych warunkach ewentualne wiązania –S-S- ulegają redukcji. Usunąć DTT poprze filtrację żelową. 3. Miareczkować obie próbki roztworem odczynnika Ellmana. Porównać wyniki.
 enzymu poddanego uprzednio. denaturacji w warunkach redukujących (w obecności merkaptoetanolu) i nieredukujących. 2. Określenie liczby wiązań –S-S- Przygotować dwie próbki enzymu w roztworze zawierającym 6 M chlorowodorek guanidyny. Poddać próbkę 1 działaniu 10 mM DTT; W tych warunkach ewentualne wiązania –S-S- ulegają redukcji. Usunąć DTT poprze filtrację żelową. 3. Miareczkować obie próbki roztworem odczynnika Ellmana. Porównać wyniki.")
28
Lokalizacja wiązań disiarczkowych
Utlenianie wiązań –S-S- kwasem nadmrówkowym Dwukierunkowa elektroforeza bibułowa Próbke białka poddaje się działaniu jodoacetamidu dla zablokowania wszystkich wolnych grup -SH Zmodyfikowane bialko poddaje się rozszczepieniu enzymatycznemu. Mieszaninę peptydów dzieli się na dwie części. Obie próbki poddaje się separacji elektroforetycznej na bibule w jednym kierunku. Rozdzielone peptydy na jednej z bibuł poddaje się działaniu par kwasu nadmrówkowego. W tych warunkach pękają wiązania disiarczkowe, a powsatłe tiole sa utleniane do reszt kwasu sulfonowego. Reszty –SH, które występowały w natywnym białku w formie wolnej, a w etapie 1 zostały zablokowane, nie ulegają w tych warunkach żadnej dalszej zmianie. Obie bibuły zostają poddane powtórnej elektroforezie, w kierunku prostopadłym do poprzedniego. Po zakończeniu elektroforezy uwidacznia się położenie peptydów w wyniku wywołania ninhydryną lub fluoreskaminą. Wyniki poddaje się analizie. Wszystkie plamki obecne na bibule, która nie była poddana działaniu kwasu nadmrówkowego powinny znajdować się na przekątnej. Jeżeli druga bibuła wyglada identycznie, to badane białko nie posiadało żadnych wiazań disiarczkowych. Wszystkie plamki znajdujące się poza przekatną na bibule poddanej działaniu kwasu nadmrówkowego odpowiadają peptydom, które były połączone wiazaniami –S-S-

29
Co zyskujemy poznając strukturę I-rzędową enzymu ?
Określenie dokładnej masy cząsteczkowej Możliwość przewidywania struktury przestrzennej białka Określenie reszt o charakterze katalitycznym poprzez związanie z odczynnikiem modyfikującym aminokwasy Interpretacja danych rendgenograficznych

30
Co zyskujemy poznając strukturę I-rzędową enzymu ?
Porównanie sekwencji łańcuchów polipeptydowych w enzymach z różnych źródeł. Pozwala to na określenie położenia tzw. reszt zakonserwowanych – mogących pełnić ważne funkcje w centrum katalitycznym lub warunkujące powstanie określonej struktury – motywu funkcyjnego Przykład: Charakterystyczna pętla wiążąca ATP (P-loop) posiada następujący wzór [AG]-X(4)-G-K-[ST]
 posiada następujący wzór. [AG]-X(4)-G-K-[ST]")
31
Określanie struktury II-rzędowej
Spektropolarymetria W metodach spektropolarymetrycznych stosuje się światło spolaryzowane, co pozwala na obserwację zjawisk powodowanych nie tylko obecnością, ale i przestrzennym ułożeniem atomów w związkach optycznie czynnych. Aktywność optyczna białek wynika z obecności kilku rodzajów asymetrii: asymetria atomów węgli a w aminokwasach asymetria helikalnych układów atomów łańcucha peptydowego i łańcuchów bocznych

32
Określanie struktury II-rzędowej
Zastosowanie spektroskopii CD do określania udziału fragmentów o określonej strukturze II-rzędowej w strukturze białka Widma CD białek o jednorodnej strukturze II-rzędowej Porównanie rzeczywistego widma CD białka z widmem skonstruowanym na podstawie przewidywania struktury II-rzędowej, którą powinien przyjąć w roztworze łańcuch polipeptydowy białka

33
Przewidywanie struktury II-rzędowej białek
Metody statystyczne/empiryczne Metody fizykochemiczne Metody mieszane

34
-helisa -kartka -zgięcie struktura nieuporządkowana
Porównanie wyników przewidywania struktury II-rzędowej białka otrzymanych przy zastosowaniu czterech różnych algorytmów obliczeniowych

35
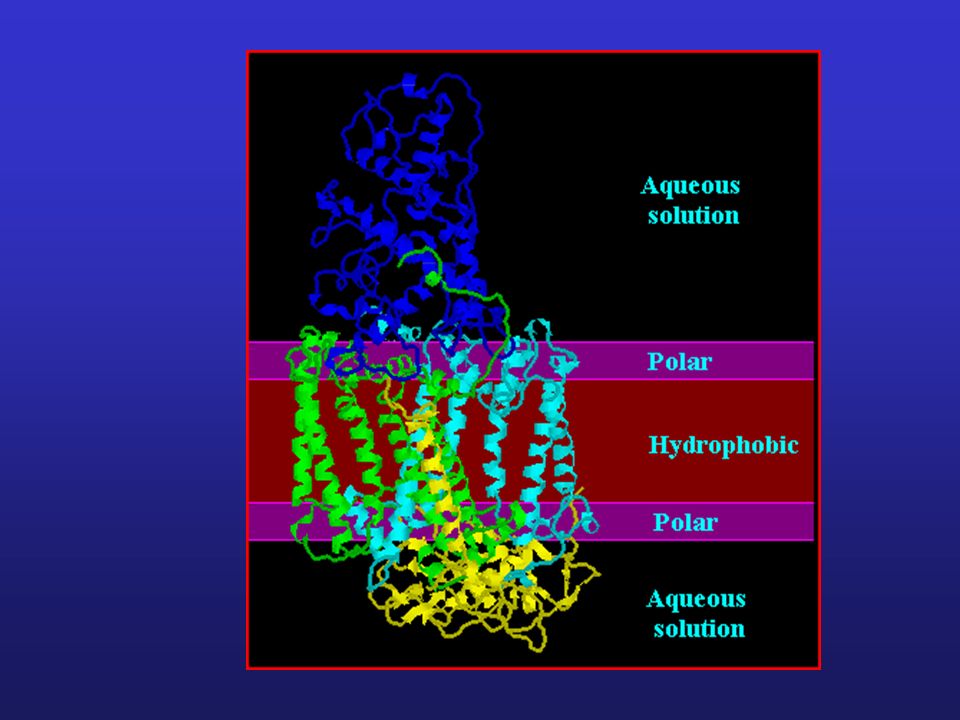
Profile hydrofobowości
Badanie białek błonowych: Identyfikacja domen hydrofobowych, fragmentów białek zakotwiczonych w błonie cytoplazmatycznej Identyfikacja fragmentów o charakterze hydrofilowym, skierowanych do cytoplazmy Przykład struktury białka błonowego

37
Parametry hydrofobowości reszt aminokwasowych
Profil hydrofobowości białka błonowego

Podobne prezentacje









, jest jednym z 20 aminokwasów.>")







>")

