Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Metody określania struktury enzymów
Enzymologia-3 Metody określania struktury enzymów

2
1. Określanie struktury I- i II-rzędowej
1.1 Określanie składu aminokwasowego i masy cząsteczkowej białka 1.2 Zastosowanie spektrometrii mas w analizie białek 1.3 Sekwencjonowanie białek 1.4 Identyfikacja wiązań disiarczkowych 1.5 Spektroskopowe metody określania struktury II-rzędowej

3
Określanie składu aminokwasowego
Hydroliza białka do składowych aminokwasów Warunki klasyczne: 6 M HCl zawierający 0.1% phenol, w próżni lub atmosferze azotu, w zamkniętej ampułce, 110 C, 18 – 96 h Problemy i ograniczenia: częściowa destrukcja reszt Ser, Thr, Tyr. Zakłada się 10% destrukcję Ser oraz 5% destrukcję Thr i Tyr w ciągu 24 h, lub przeprowadza się hydrolizę trzech próbek, odpowiednio przez 24, 48 i 72 h i następnie ekstrapoluje się wyniki dla momentu zero reszty Asn i Gln są przekształcane w Asp i Glu. Białko poddaje się działaniu bis (1,1-trifluoro-acetoksy)jodobenzenu /36 mg/ml in 0.01 M TFA, 4 h, 60 C, w ciemności/. W tych warunkach reszty karboksyamidowe są przekształcane w aminy całkowita destrukcja Trp Konieczne przeprowadzenie osobnej hydrolizy. Warunki: 3 M kwas p-toluenosulfonowy, 110 C, 24 – 72 h, w prózni
jodobenzenu /36 mg/ml in 0.01 M TFA, 4 h, 60 C, w ciemności/. W tych warunkach reszty karboksyamidowe są przekształcane w aminy. całkowita destrukcja Trp. Konieczne przeprowadzenie osobnej hydrolizy. Warunki: 3 M kwas p-toluenosulfonowy, 110 C, 24 – 72 h, w prózni.")
4
Separacja i analiza mieszanin aminokwasów
Separacja mieszaniny PTC-aminokwasów metodą HPLC. Wypełnienie: octadecylosilan, elucja mieszaniną acetonitryl /woda Chromatografia jonowymienna mieszaniny aminokwasów. Kolumna Dowex 50WX4 Derywatyzacja aminokwasów Analizatory jonowymienne – po kolumnie Ninhydryna; barwne pochodne, detekcja przy 570 nm lub 440 nm (Pro); fluoreskamina lub aldehyd ftalowy + 2-tioetanol; produkty fluoryzujące; HPLC w układzie faz odwróconych – przed kolumną fenylotioizocyjanian; PTC-pochodne chlorek dansylu; DNS-pochodne chlorek N-(9-fluorenylometoksykarbonylu; FMOC-pochodne
; fluoreskamina lub aldehyd ftalowy + 2-tioetanol; produkty. fluoryzujące; HPLC w układzie faz odwróconych – przed kolumną. fenylotioizocyjanian; PTC-pochodne. chlorek dansylu; DNS-pochodne. chlorek N-(9-fluorenylometoksykarbonylu; FMOC-pochodne.")
5
Metody określania masy cząsteczkowej białek
Metody bezpośrednie: sedymentacja (wirowanie strefowe) spektrometria mas (MS ESI) Metody porównawcze: elektroforeza w żelu poliakrylamidowym (SDS-PAGE i natywna) chromatografia rozmiarów wykluczających
 spektrometria mas (MS ESI) Metody porównawcze: elektroforeza w żelu poliakrylamidowym (SDS-PAGE i natywna) chromatografia rozmiarów wykluczających.")
6
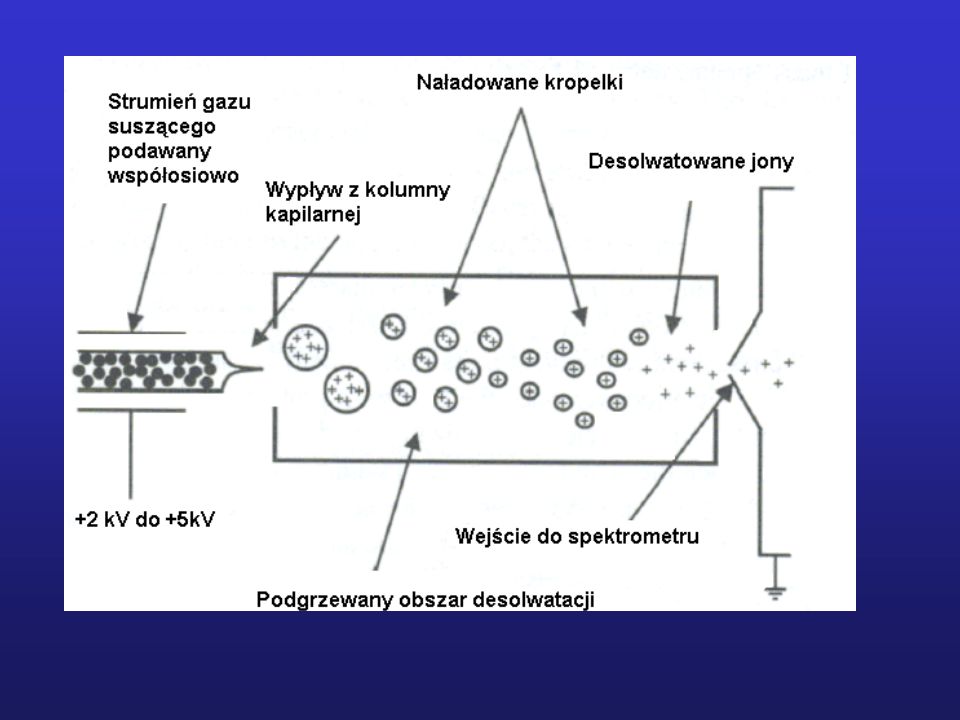
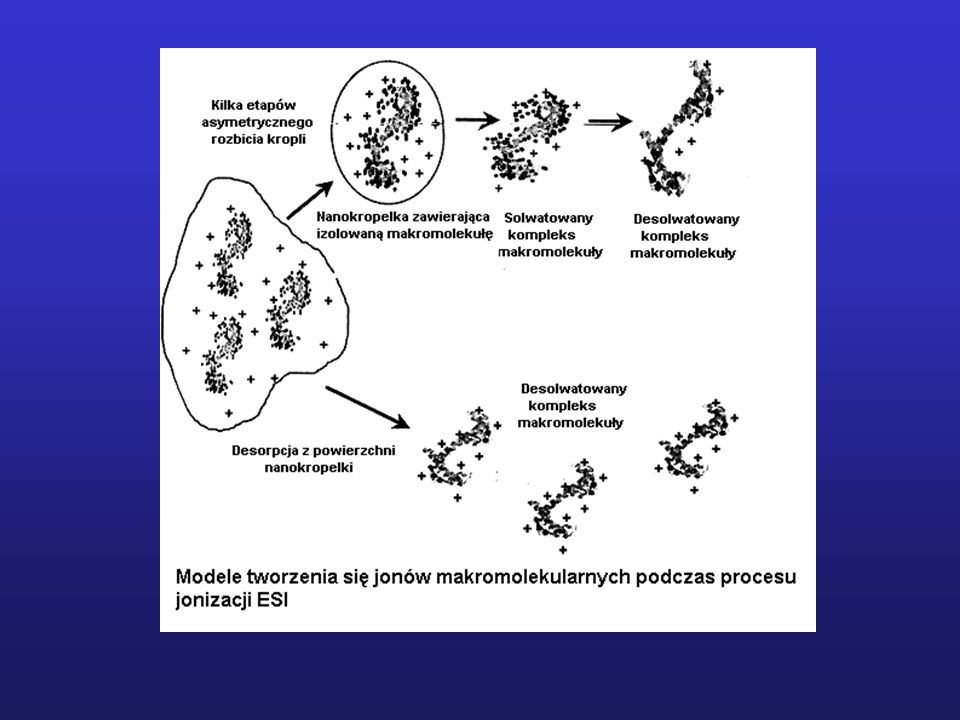
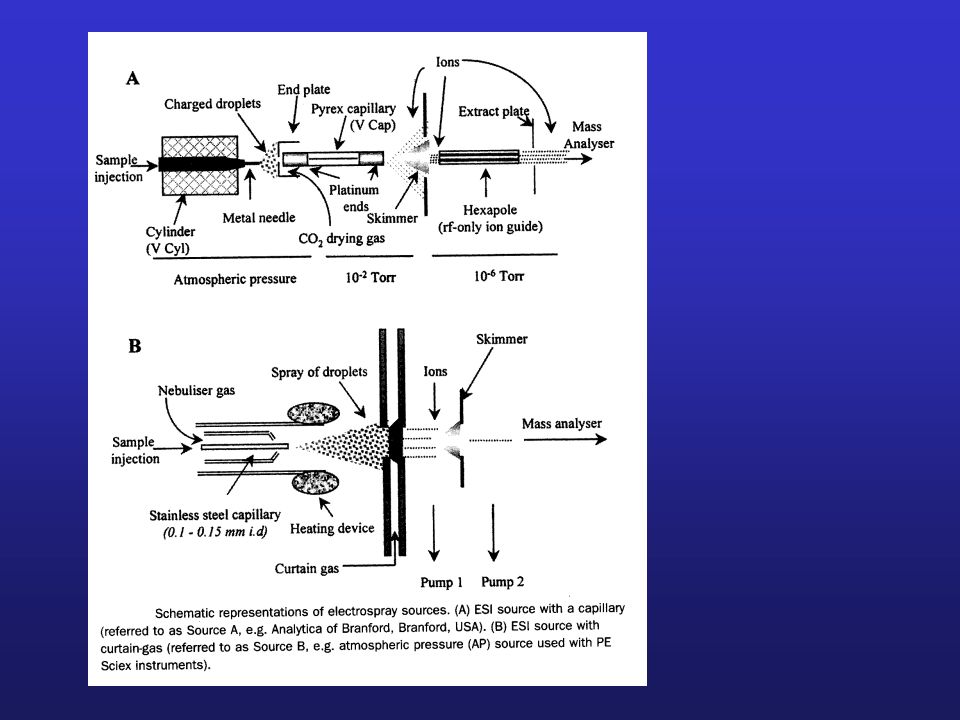
Schemat blokowy spektrometru mas

10
Schemat spektrometru MALDI-TOF

11
Sekwencjonowanie białek
Rozszczepienie łańcucha białka na fragmenty metodami enzymatycznymi lub chemicznymi Separacja i izolacja peptydów Sekwencjonowanie peptydów Określenie kompletnej sekwencji łańcuch białka na podstawie porównania nakładających się sekwencji peptydów

12
Enzymatyczne metody rozszczepienia białek
Zasady ogólne: białka należy zdenaturować przed podddaniem ich trawieniu enzymatycznemu należy używać lotnych buforów (można je usunąć poprze liofilizację) należy używać enzymów proteolitycznych specjalnej jakości (sequencing grade) warunki reakcji powinny być zoptymalizowane Enzymy wykorzystywane do specyficznego cięcia: Trypsyna – karboksylowa strona reszt Arg, Lys i S-aminoetylocysteiny; specyficzność można ograniczyć do Arg poprzez modyfikacje reszt lizyny np, bezwodnikiem kwasu bursztynowego; Chymotrypsyna – karboksylowa strona Phe, Trp, Tyr and Leu (w kolejności zmniejszającej się podatności) Termolizyna – aminowa strona Leu, Ile, Phe, Val Proteaza V8 ze S. aureus – wiązania Glu-X oraz (znacznie mniej efektywnie) Asp-X Endoproteinaza Lys-C – wiązania Lys-X
 należy używać enzymów proteolitycznych specjalnej jakości (sequencing grade) warunki reakcji powinny być zoptymalizowane. Enzymy wykorzystywane do specyficznego cięcia: Trypsyna – karboksylowa strona reszt Arg, Lys i S-aminoetylocysteiny; specyficzność. można ograniczyć do Arg poprzez modyfikacje reszt lizyny np, bezwodnikiem. kwasu bursztynowego; Chymotrypsyna – karboksylowa strona Phe, Trp, Tyr and Leu (w kolejności zmniejszającej. się podatności) Termolizyna – aminowa strona Leu, Ile, Phe, Val. Proteaza V8 ze S. aureus – wiązania Glu-X oraz (znacznie mniej efektywnie) Asp-X. Endoproteinaza Lys-C – wiązania Lys-X.")
13
Określanie masy cząsteczkowej białka z widma MS ESI
Zasada obliczania masy cząsteczkowej A/ Dla dwóch sąsiednich pików jonizacyjnych m1 i m2, gdzie m2>m1 m1 = (M + z1)/z1 m2 = [(M + (z1 – 1)]/(z1 – 1) B/ Podstawiając za m1 i m2 wartości m/z sąsiednich pików, rozwiązujemy układ równań względem niewiadomych M i z1
/z1. m2 = [(M + (z1 – 1)]/(z1 – 1) B/ Podstawiając za m1 i m2. wartości m/z sąsiednich pików, rozwiązujemy układ równań. względem niewiadomych M i z1.")
14
Metody chemicznego cięcia łańcuchów białkowych
1. Indukowane działaniem CNBr rozszczepienie przy resztach Met

15
2. Rozszczepienie wiązań Asn-Gly działaniem N-hydroksyloaminy
Warunki : 6 M chlorowodorek guanidyny, 2 M chlorowodorek hydroksyloaminy, pH = 9.0, w obecności 4.5 M LiOH

16
Sekwencjonowanie białek
Degradacja Edmana N C S + H 2 R 1 O pH 8 F 3 bezwodny rozcieńczony kwas mineralny fenylotiohydantoina (PTH) n-peptyd (n-1)-peptyd kolejny PITC cykl (DABITC) (DNS) DNS-pochodne aminokwasów można wykryć na poziome fentomolowym Metoda DABITC/PITC DABITC tworzy barwne pochodne z aminokwasami; wysoka czułość, ale tylko 60% efektywność sprzęgania. Metoda DABITC/PITC – każdy cykl składa się z dwóch etapów sprzęgania: DABITC, a następnie PITC
 n-peptyd. (n-1)-peptyd. kolejny. PITC. cykl. (DABITC) (DNS) DNS-pochodne aminokwasów. można wykryć na poziome. fentomolowym. Metoda DABITC/PITC. DABITC tworzy barwne pochodne z aminokwasami; wysoka czułość, ale tylko 60% efektywność sprzęgania. Metoda DABITC/PITC – każdy cykl składa się z dwóch. etapów sprzęgania: DABITC, a następnie PITC.")
17
Mikrosekwencjonowanie peptydów
Możliwość analizy próbek w ilościach nanogramowych

18
Zastosowanie spektrometrii mas do analizy peptydów
1. MS-FAB (Fast Atom Bombardment) Materiał analizowany: peptydy powstające w wyniku rozszczepienia chemicznego lub enzymatycznego, rozpuszczone w 30% kwasie octowym (0.5 – 1 g/l); taki roztwór miesza się z glicerolem (50:50, v/v), Próbka nie może zawierać soli Na próbkę działa się działa się strumieniem atomów Ar lub Xe lub jonami Cs+ (5 – 10 kV); Selekcja jonów: analizator sektorów magnetycznych lub TOF 2. MS MALDI-TOF Materiał analizowany: Mieszaniny peptydów powstające w wyniku traktowania białka kilkoma różnymi enzymami proteolitycznymi (osobne próbki). Zwykle wstępnie blokuje się reszty cysteiny i/lub lizyny. Próbka w matrycy stałej. Złożone widma analizowane są w celu identyfikacji poszczególnych peptydów. Metodę można także stosować do identyfikacji miejsc modyfikacji białek
 Materiał analizowany: peptydy powstające w wyniku rozszczepienia chemicznego. lub enzymatycznego, rozpuszczone w 30% kwasie octowym (0.5 – 1 g/l); taki roztwór. miesza się z glicerolem (50:50, v/v), Próbka nie może zawierać soli. Na próbkę działa się działa się strumieniem atomów Ar lub Xe lub jonami. Cs+ (5 – 10 kV); Selekcja jonów: analizator sektorów magnetycznych lub TOF. 2. MS MALDI-TOF. Materiał analizowany: Mieszaniny peptydów powstające w wyniku traktowania białka. kilkoma różnymi enzymami proteolitycznymi (osobne próbki). Zwykle wstępnie blokuje. się reszty cysteiny i/lub lizyny. Próbka w matrycy stałej. Złożone widma analizowane są w celu identyfikacji poszczególnych peptydów. Metodę można także stosować do identyfikacji miejsc modyfikacji białek.")
19
MS-FAB w analizie peptydów
Widmo MS FAB peptydu Arg-Pro-Val-Trp-Pro- Asn-Gly-Ala-Glu-Ser-Ala-Glu-Ala-Phe-Pro-Leu-Glu-Phe. Mniejszy rysunek – powiększenie obszaru (M + H)+ Sekwencjonowanie białek z zastosowaniem tandemowej spektrometrii MS-FAB
+ Sekwencjonowanie białek z zastosowaniem tandemowej. spektrometrii MS-FAB.")
20
Analiza MS MALDI-TOF mieszanin peptydów otrzymanych w wyniku działania chymotrypsyny
na enzym, syntazę GLcN-6-P: A – forma natywna; B – forma poddana działaniu inaktywatora lączącego się z enzymem wiązaniem kowalencyjnym Wynik nałożenia obszarów 200 – 800 widm A i B. Sygnałów obecnych w obu widmach nie pokazano

21
Identyfikacja wiązań disiarczkowych
1. Czy enzym zawiera w swojej strukturze wiązania disiarczkowe? Należy porównać ruchliwość elektroforetyczną (SDS-PAGE) enzymu poddanego uprzednio Denaturacji w warunkach redukujących (w obecności merkaptoetanolu) i nieredukujących 2. Określenie liczby wiązań –S-S- Przygotować dwie próbki enzymu w roztworze zawierającym 6 M chlorowodorek guanidyny Poddać próbkę 1 działaniu 10 mM DTT; W tych warunkach ewentualne wiązania –S-S- ulegają redukcji. Usunąć DTT poprze filtrację żelową. 3. Miareczkować obie próbki roztworem odczynnika Ellmana. Porównać wyniki.
 enzymu poddanego uprzednio. Denaturacji w warunkach redukujących (w obecności merkaptoetanolu) i nieredukujących. 2. Określenie liczby wiązań –S-S- Przygotować dwie próbki enzymu w roztworze zawierającym 6 M chlorowodorek guanidyny. Poddać próbkę 1 działaniu 10 mM DTT; W tych warunkach ewentualne wiązania –S-S- ulegają redukcji. Usunąć DTT poprze filtrację żelową. 3. Miareczkować obie próbki roztworem odczynnika Ellmana. Porównać wyniki.")
22
Lokalizacja wiązań disiarczkowych
Utlenianie wiązań –S-S- kwasem nadmrówkowym Dwukierunkowa elektroforeza bibułowa Próbke białka poddaje się działaniu jodoacetamidu dla zablokowania wszystkich wolnych grup -SH Zmodyfikowane bialko poddaje się rozszczepieniu enzymatycznemu. Mieszaninę peptydów dzieli się na dwie części. Obie próbki poddaje się separacji elektroforetycznej na bibule w jednym kierunku. Rozdzielone peptydy na jednej z bibuł poddaje się działaniu par kwasu nadmrówkowego. W tych warunkach pękają wiązania disiarczkowe, a powsatłe tiole sa utleniane do reszt kwasu sulfonowego. Reszty –SH, które występowały w natywnym białku w formie wolnej, a w etapie 1 zostały zablokowane, nie ulegają w tych warunkach żadnej dalszej zmianie. Obie bibuły zostają poddane powtórnej elektroforezie, w kierunku prostopadłym do poprzedniego. Po zakończeniu elektroforezy uwidacznia się położenie peptydów w wyniku wywołania ninhydryną lub fluoreskaminą. Wyniki poddaje się analizie. Wszystkie plamki obecne na bibule, która nie była poddana działaniu kwasu nadmrówkowego powinny znajdować się na przekątnej. Jeżeli druga bibuła wyglada identycznie, to badane białko nie posiadało żadnych wiazań disiarczkowych. Wszystkie plamki znajdujące się poza przekatną na bibule poddanej działaniu kwasu nadmrówkowego odpowiadają peptydom, które były połączone wiazaniami –S-S-

23
Określanie struktury II-rzędowej
Zastosowanie spektroskopii CD do określania udziału fragmentów o określonej strukturze II-rzędowej w strukturze białka Widma CD białek o jednorodnej strukturze II-rzędowej Porównanie rzeczywistego widma CD białka z widmem skonstruowanym na podstawie przewidywania struktury II-rzędowej, którą powinien przyjąć w roztworze łańcuch polipeptydowy białka

24
-helisa -kartka -zgięcie struktura nieuporządkowana
Porównanie wyników przewidywania struktury II-rzędowej białka otrzymanych przy zastosowaniu czterech różnych algorytmów obliczeniowych

25
Parametry hydrofobowości reszt aminokwasowych
Przykład struktury białka błonowego Parametry hydrofobowości reszt aminokwasowych Profil hydrofobowości białka błonowego

Podobne prezentacje









, jest jednym z 20 aminokwasów.>")




>")




