Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Chemia fizyczna Termodynamika 2013/14

2
Wiadomości organizacyjne
Tadeusz Hofman, Zakład Chemii Fizycznej, p. 148, Gmach Chemii Materiały internetowe: Konsultacje Egzamin środa 13:15-14:00 23.06 poniedziałek 8:15-10:00 AZ, 350AB 26.06 czwartek 1.09 15:15-17:00

3
Regulamin (1) 1. Warunkiem dopuszczenia do egzaminu jest zaliczenie ćwiczeń rachunkowych. 2. Ocena z Ćwiczeń - bdb zwalnia z egzaminu i przepisywana jest jako egzaminacyjna. 3. Egzamin składa się z dwugodzinnej części pisemnej, na którą składa się w połowie materiał z Termodynamiki i w połowie z Kinetyki i Elektrochemii oraz ustnej (zwykle następnego dnia). 4. Warunkiem zaliczenia egzaminu jest niezależne zaliczenie obu jego części. Można to zrobić w różnych terminach (np. zaliczona termodynamika w pierwszym terminie zostanie uznana podczas kolejnego egzaminu), ale pod warunkiem zdania całego egzaminu w sesji letniej lub jesiennej tego samego roku. 5. Przewiduje się 3 terminy egzaminów (dwa w sesji letniej i jeden w sesji jesiennej).
. 4. Warunkiem zaliczenia egzaminu jest niezależne zaliczenie obu jego części. Można to zrobić w różnych terminach (np. zaliczona termodynamika w pierwszym terminie zostanie uznana podczas kolejnego egzaminu), ale pod warunkiem zdania całego egzaminu w sesji letniej lub jesiennej tego samego roku. 5. Przewiduje się 3 terminy egzaminów (dwa w sesji letniej i jeden w sesji jesiennej).")
4
Regulamin (2) 6. Student ma prawo do dwukrotnego poprawiania niezdanej połówki egzaminu, ale tylko w ramach terminów ustalonych przed sesją egzaminacyjną. 7. W zależności od liczby punktów zdobytych (P) na egzaminie pisemnym wynikają następujące konsekwencje: P < 40% - nzal 40% < P < 50% - egzamin ustny P > 50% - egzamin cząstkowy jest zdany; ocena łączna jest średnią arytmetyczną wyników z termodynamiki oraz kinetyki i elektrochemii. Ocena wynika z powyższego zestawienia, ale można ją poprawiać na egzaminie ustnym. 8. Jedyne materiały dopuszczalne na egzaminie pisemnym to: - kalkulator, - przyrząd do pisania, - linijka, - kartka papieru milimetrowego (do rysowania wykresów).
 na egzaminie pisemnym wynikają następujące konsekwencje: P < 40% - nzal 40% < P < 50% - egzamin ustny P > 50% - egzamin cząstkowy jest zdany; ocena łączna jest średnią arytmetyczną wyników z termodynamiki oraz kinetyki i elektrochemii. Ocena wynika z powyższego zestawienia, ale można ją poprawiać na egzaminie ustnym. 8. Jedyne materiały dopuszczalne na egzaminie pisemnym to: - kalkulator, - przyrząd do pisania, - linijka, - kartka papieru milimetrowego (do rysowania wykresów).")
5
Regulamin (3) Konsekwencją posiadania przy sobie w trakcie egzaminu innych materiałów, a szczególnie ściąg i podobnych pomocy, jest niezaliczenie egzaminu. Podkreślam – posiadania, a niekoniecznie korzystania. 9.1. Egzamin pisemny składa się z 5 pytań i 1 zadania. Pytania wybierane są z zestawu około 60 problemów. Pytania pogrubione, tak zwane NIEZAPOMINAJKI, oceniane są dwa razy wyżej niż pytania pozostałe. 9.2. Egzamin ustny traktowany jest jako poprawa egzaminu pisemnego. Warunkiem koniecznym jego zaliczenia, jest udzielenie poprawnych odpowiedzi na wszystkie niezapominajki, które nie zostały zaliczone podczas egzaminu pisemnego. Druga część będzie polegać na odpowiedzi (pisemnej + ewentualne ustne wyjaśnienia) na kilka wylosowanych pytań z podanej listy. Nie muszą to być pytania z egzaminu pisemnego. Na egzaminie ustnym można również poprawiać ocenę zaliczającą. W tym przypadku pytania mogą wykraczać poza listę pytań egzaminacyjnych.
 na kilka wylosowanych pytań z podanej listy. Nie muszą to być pytania z egzaminu pisemnego. Na egzaminie ustnym można również poprawiać ocenę zaliczającą. W tym przypadku pytania mogą wykraczać poza listę pytań egzaminacyjnych.")
6
Egzamin - pytania 1. Podać definicje: funkcji stanu, różniczki zupełnej, parametrów intensywnych i ekstensywnych (przykłady parametrów). 2. Definicje procesów: adiabatycznego, diatermicznego, kwazystatycznego (przykłady). 4. I Zasada Termodynamiki; definicja entalpii; I Zasada wyrażona poprzez entalpię. 9. Podać definicję standardowej entalpii tworzenia (spalania) acetonu [*] (CH3COCH3(c)) w temperaturze T; Obliczyć standardową entalpię reakcji: CH3OH(g) + CO(g) → CH3COOH(g) [*] wykorzystując standardowe entalpie tworzenia (spalania).
. 2. Definicje procesów: adiabatycznego, diatermicznego, kwazystatycznego (przykłady). 4. I Zasada Termodynamiki; definicja entalpii; I Zasada wyrażona poprzez entalpię. 9. Podać definicję standardowej entalpii tworzenia (spalania) acetonu [*] (CH3COCH3(c)) w temperaturze T; Obliczyć standardową entalpię reakcji: CH3OH(g) + CO(g) → CH3COOH(g) [*] wykorzystując standardowe entalpie tworzenia (spalania).")
7
Egzamin – przykładowe zadanie
Prężności par nasyconych nad czystymi składnikami A i B, mogą być wyrażone w postaci: gdzie i = A, B w interesującym nas przedziale temperatur. Mieszanina ciekła A + B jest praktycznie roztworem doskonałym. Podać i naszkicować orientacyjnie równania izobary (dla p = p0) równowagi ciecz-para dla tego układu czyli temperatury wrzenia w funkcji składu fazy ciekłej i składu fazy gazowej.
 równowagi ciecz-para dla tego układu czyli temperatury wrzenia w funkcji składu fazy ciekłej i składu fazy gazowej.")
8
http://www.ch.pw.edu.pl/~hof/bio.htm Literatura
Chemia fizyczna, praca zbiorowa, PWN, Warszawa 1980. P.W. Atkins, Chemia fizyczna, PWN, Warszawa 2001. H. Buchowski, W. Ufnalski, Podstawy termodynamiki, WNT, Warszawa 1994. H. Buchowski, W. Ufnalski, Gazy, ciecze, płyny, WNT, Warszawa 1994. H. Buchowski, W. Ufnalski, Roztwory, WNT, Warszawa 1995. H. Buchowski, W. Ufnalski, Równowagi chemiczne. WNT, Warszawa 1995

9
Literatura K. Gumiński, Termodynamika, PWN, Warszawa 1974. K. Pigoń, K. Ruziewicz, Chemia fizyczna. Podstawy fenomenologiczne. PWN, Warszawa, 2005. K. Zalewski, Wykłady z mechaniki i termodynamiki statystycznej dla chemików, PWN, Warszawa 1982. K. Zalewski, Wykłady z termodynamiki fenomenologicznej i statystycznej, PWN, Warszawa 1978.

10
Czym zajmuje się termodynamika?

11
Czym zajmuje się termodynamika?
Termodynamika zajmuje się opisem (mechanicznym) układów makroskopowych, złożonych z olbrzymiej liczby elementów składowych (cząsteczek). wieloelementowość!
 układów makroskopowych, złożonych z olbrzymiej liczby elementów składowych (cząsteczek). wieloelementowość!")
12
Jak znaleźć stan mechaniczny układu cząsteczek – równania Newtona
Układ 3 równań różniczkowych drugiego rzędu Ale czy rzeczywiście taka liczba parametrów jest niezbędna ? Do rozwiązania niezbędne jest 6 warunków brzegowych Dla N cząsteczek – 6N parametrów! Parametry makroskopowe są uśrednionymi i zsumowanymi parametrami cząsteczkowymi

13
Podsumowanie zadań termodynamiki
Cel - znalezienie (i wykorzystywanie w celach praktycznych) związków pomiędzy parametrami makroskopowymi dla pewnych stanów oraz procesów. Dwie drogi: Termodynamika statystyczna – wyprowadza związki na podstawie właściwości cząsteczkowych. Termodynamika klasyczna (fenomenologiczna) – opiera się na czterech aksjomatach zwanych Zasadami Termodynamiki i nie odwołuje się do cząsteczkowej struktury materii.
 związków pomiędzy parametrami makroskopowymi dla pewnych stanów oraz procesów. Dwie drogi: Termodynamika statystyczna – wyprowadza związki na podstawie właściwości cząsteczkowych. Termodynamika klasyczna (fenomenologiczna) – opiera się na czterech aksjomatach zwanych Zasadami Termodynamiki i nie odwołuje się do cząsteczkowej struktury materii.")
14
Parametry termodynamiczne opisujące układ

15
Parametry termodynamiczne
V, n1, n2, …., p, T parametry intensywne: p, T - ciśnienie, temperatura parametry ekstensywne: V, n1, n2, …. – objętość, ilości składników (liczby moli) p, T, V, n1, n2, …. T
 p, T, V, n1, n2, …. T.")
16
Funkcje (parametry) stanu – co wynika z prostej konstatacji?
Jeśli F jest funkcją stanu, to jest ona bezpośrednio całkowalna dF jest różniczką zupełną, tzn. dla funkcji F(x1,x2,...,xn) Kolejność różniczkowania drugich mieszanych pochodnych cząstkowych jest dowolna, czyli spełnione są relacje Maxwella dla każdej pary i,j
 Kolejność różniczkowania drugich mieszanych pochodnych cząstkowych jest dowolna, czyli spełnione są relacje Maxwella. dla każdej pary i,j.")
17
układ otoczenie

18
układ izolowany przepływ masy układ zamknięty układ otwarty
masa, energia osłona układ izolowany przepływ masy układ zamknięty układ otwarty

19
Osłony A jest w równowadze z C
osłona adiabatyczna - taka, że tylko procesy w otoczeniu związane z wykonywaniem pracy wpływają na stan układu → proces adiabatyczny. osłona diatermiczna (termicznie przewodząca) - taka, że dla trzech układów (A, B, C) ograniczonych taką osłoną, spełniona jest następująca relacja A jest w równowadze z B B w równowadze z C A jest w równowadze z C C’ A B C
 - taka, że dla trzech układów (A, B, C) ograniczonych taką osłoną, spełniona jest następująca relacja. A jest w równowadze z B. B w równowadze z C. A jest. w równowadze z C. C’ A. B. C.")
20
„Przechodniość” w życiu
? Kuba Lukrecja Lukrecja Ignacy

21
Zerowa Zasada Termodynamiki
Jest równoważna postulatowi istnienia osłony diatermicznej Z warunku równowagi i własności osłony diatermicznej wynikają związki pomiędzy parametrami układów będących w stanie równowagi Co jest możliwe tylko wtedy, kiedy istnieje wspólny parametr Jest to definicja temperatury (empirycznej)
")
22
Proces odwracalny (kwazystatyczny)
p - dp p + dp pz Proces odwracalny (kwazystatyczny) - taki, że nieskończenie mała zmiana wartości parametrów wystarczy do odwrócenia jego kierunku. Zmiana ciśnienia o 2·dp prowadzi do odwrócenia procesu.
 - taki, że nieskończenie mała zmiana wartości parametrów wystarczy do odwrócenia jego kierunku. Zmiana ciśnienia o 2·dp prowadzi do odwrócenia procesu.")
23
Zasada Duhema (na razie empiryczna)
W jednofazowym układzie zamkniętym (n=const) dwa parametry wystarczą do pełnego opisu stanu układu. f(p,V, T) = równanie stanu Najprostsze równanie stanu: równanie stanu gazu doskonałego: pV = nRT
 dwa parametry wystarczą do pełnego opisu stanu układu. f(p,V, T) = 0 - równanie stanu. Najprostsze równanie stanu: równanie stanu gazu doskonałego: pV = nRT.")
24
Praca objętościowa dl V p pz czy ?

25
Praca objętościowa – jaki znak?
Patrzymy na Świat z perspektywy Układu Praca wykonana przez układ nad otoczeniem układ traci energię praca powinna być ujemna dV > dw > 0

26
Praca odwracalna jest minimalna (maksymalna)
Praca objętościowa p pz pz p proces odwracalny dw -pzdV -pdV min szybkość maks Praca odwracalna jest minimalna (maksymalna)
")
27
Inne rodzaje pracy

28
Czy praca objętościowa jest funkcją stanu?
W ogólnym przypadku nie (!), bo dw = - pzdV A dla przemiany odwracalnej? dw = - pdV
, bo. dw = - pzdV. A dla przemiany odwracalnej dw = - pdV.")
29
Czy praca objętościowa jest funkcją stanu?
T=const Praca objętościowa nie jest funkcją stanu także dla przemiany odwracalnej! B V

30
Podsumowanie i wnioski
Termodynamika zajmuje się układami makroskopowymi (złożonymi z olbrzymiej liczby cząsteczek). Opisuje je za pomocą parametrów (funkcji) stanu. Dwa sposoby znajdywania związków pomiędzy parametrami – termodynamika klasyczna i statystyczna. Temperatura zdefiniowana poprzez Zerową Zasadę Termodynamiki. Praca objętościowa jako konsekwencja przyjęcia parametru V.
. Opisuje je za pomocą parametrów (funkcji) stanu. Dwa sposoby znajdywania związków pomiędzy parametrami – termodynamika klasyczna i statystyczna. Temperatura zdefiniowana poprzez Zerową Zasadę Termodynamiki. Praca objętościowa jako konsekwencja przyjęcia parametru V.")
31
Podsumowanie i wnioski
Praca objętościowa nie jest funkcją stanu, w związku z czym nie jest spełniana fundamentalna (!) równość: Jedyna możliwość „uratowania” zasady zachowania energii to przyjęcie istnienia jeszcze innego sposobu przekazywania energii (Q). Wtedy byłoby
 równość: Jedyna możliwość „uratowania zasady zachowania energii to przyjęcie istnienia jeszcze innego sposobu przekazywania energii (Q). Wtedy byłoby.")
32
I ZASADA TERMODYNAMIKI
Postuluje się istnienie funkcji stanu, zwanej energią wewnętrzną (U), która ma następujące właściwości: 1. Jest funkcją ekstensywną 2. Jej różniczka zupełna równa się różniczkowej pracy w przemianie adiabatycznej w układzie zamkniętym dU = (dw)ad
, która ma następujące właściwości: 1. Jest funkcją ekstensywną. 2. Jej różniczka zupełna równa się różniczkowej pracy w przemianie adiabatycznej w układzie zamkniętym. dU = (dw)ad.")
33
Ciepło definicja ciepła Bilans energii

34
Jest funkcją ekstensywną!
Entalpia Jest funkcją ekstensywną! Bilans entalpii

35
Ciepło jako funkcja stanu
Germain Hess ( ) Prawo Hessa
 Prawo Hessa.")
36
Jak mierzymy efekt cieplny?
Pojemność cieplna pod stałym ciśnieniem Pojemność cieplna w stałej objętości

37
Termochemia ENTALPIA

38
Standardowa entalpia reakcji (ΔHo)
N2 + 3H2 → 2NH3 Niejednoznaczność zapisu! Konieczność ścisłego zdefiniowania stanu początkowego i końcowego!

39
Standardowa entalpia reakcji (ΔHo) – reakcja standardowa
Reakcja biegnie do końca. Bierze w niej udział liczba moli reagentów wynikająca z równania stechiometrycznego. Temperatura oraz ciśnienie w stanie początkowym (substraty) i końcowym (produkty) są takie same. Reagenty występują w stanach standardowych.
 i końcowym (produkty) są takie same. Reagenty występują w stanach standardowych.")
40
Standardowa entalpia reakcji (ΔHo) – stan standardowy
Ciśnienie p° = 1 bar, gazy (też mieszaniny) - czyste gazy doskonałe, substancje skondensowane (czyste lub w roztworze, poza jonami) - czyste składniki, jony w roztworze - roztwór doskonały o stężeniu 1 mol/ 1000 g rozpuszczalnika.
 - czyste gazy doskonałe, substancje skondensowane (czyste lub w roztworze, poza jonami) - czyste składniki, jony w roztworze - roztwór doskonały o stężeniu 1 mol/ 1000 g rozpuszczalnika.")
41
Standardowa entalpia reakcji (ΔHo) – problem wyznaczenia
N2 + 3H2 → 2NH3 ΔHo = suma entalpii produktów – suma entalpii substratów ? Jest to niewykonalne, ponieważ nie wyznaczymy H reagentów! Możemy jedynie posługiwać się zmianami entalpii. Zadanie: zdefiniować jakąś podstawową, ogólną reakcję, której standardowe entalpie stanowiłyby podstawę obliczania standardowych entalpii dowolnej reakcji.

42
Standardowa entalpia tworzenia (ΔHf o)
Jest to standardowa entalpia następującej reakcji: pierwiastki w stanach termodynamicznie trwałych 1 mol związku dla C2H5OH(c) (T = 300 K)? dla 2C(grafit) + 3H2(g) + 1/2O2(g)→ C2H5OH(c)
 (T = 300 K) dla 2C(grafit) + 3H2(g) + 1/2O2(g)→ C2H5OH(c)")
43
Uogólnione współczynniki stechiometryczne
N2 + 3H2 → 2NH3 Współczynniki stechiometryczne? 1, 3, 2? Nie! , -3, +2 ΔHo = suma entalpii produktów – suma entalpii substratów

44
Standardowa entalpia z entalpii tworzenia
pierwiastki w stanach termodynamicznie trwałych w ilościach wynikających ze stechiometrii

45
Standardowa entalpia z entalpii tworzenia

46
Standardowa entalpia spalania (ΔHsp o)
Jest to standardowa entalpia następującej reakcji: 1 mol związku + nO2(g) mCO2(g) + kH2O
 mCO2(g) + kH2O.")
47
Zależność ∆H od temperatury – prawo Kirchhoffa
Związki między parametrami zdefiniowane poprzez pochodne! Gustav Kirchhoff ( )
")
48
Standardowa energia wewnętrzna (ΔUo)
O różnicy pomiędzy standardową entalpią a energią wewnętrzną decyduje zmiana objętości.

49
Standardowa energia wewnętrzna (ΔUo) - przykład
N2(g) + 3H2(g) → 2NH3(g) N2(g) + 3H2(g) → 2NH3(c)
 + 3H2(g) → 2NH3(g) N2(g) + 3H2(g) → 2NH3(c)")
50
Średnia termochemiczna energia wiązań EXY
….X-Y…(g) X(g)+ Y(g) Przykład: CH4(g) + 4Cl2(g) → CCl4(c) + 4HCl(g)
 X(g)+ Y(g) Przykład: CH4(g) + 4Cl2(g) → CCl4(c) + 4HCl(g)")
51
Jeszcze parę słów o temperaturze
Czego temperaturę mierzymy? Mierzymy temperaturę termometru! Musi istnieć wspólny parametr. Wtedy temperatura termometru jest równa temperaturze układu. Zapewnia nam to istnienie osłony diatermicznej, co gwarantuje Zerowa Zasada Termodynamiki.

52
Jak zmierzyć temperaturę?
Jeśli nie bezpośrednio, to jak? Równanie stanu F(T, p, V, n = const) = 0. Stąd Ścisłą zależność daje nam pochodna: Ale pochodnej tej nie znamy! Co robić? Najprostsze rozwiązanie: Potrzebne dwa punkty do kalibracji!
 = 0. Stąd. Ścisłą zależność daje nam pochodna: Ale pochodnej tej nie znamy! Co robić Najprostsze rozwiązanie: Potrzebne dwa punkty do kalibracji!")
53
Jeszcze parę słów o temperaturze
Wady „takiej” temperatury: arbitralność definicji, uzależnienie od cieczy, termometrycznej. Anders Celsius ( )
")
54
Jeszcze parę słów o temperaturze
Różne gazy, p 0, m = const pV t/ oC t =-273,15

55
Jeszcze parę słów o temperaturze
Różne gazy, p 0, V0 (T0,p0) = const pV William Thomson ( ) t/ oC t =-273,15 skala Kelvina
 = const. pV. William Thomson ( ) t/ oC. t =-273,15. skala Kelvina.")
56
Jeszcze parę słów o temperaturze
Termometr gazowy i temperatura empiryczna

57
I ZASADA TERMODYNAMIKI
Postuluje się istnienie funkcji stanu, zwanej energią wewnętrzną (U), która ma następujące właściwości: 1. Jest funkcją ekstensywną 2. Jej różniczka zupełna równa się różniczkowej pracy w przemianie adiabatycznej w układzie zamkniętym dU = (dw)ad
, która ma następujące właściwości: 1. Jest funkcją ekstensywną. 2. Jej różniczka zupełna równa się różniczkowej pracy w przemianie adiabatycznej w układzie zamkniętym. dU = (dw)ad.")
58
Dlaczego pewne procesy zachodzą, a inne nie?
W świecie, w którym żyjemy zachodzą tylko niektóre procesy, które nie są sprzeczne z I Zasadą. I Zasada nie wystarczy! Te procesy, które zachodzą, są nieodwracalne. niektóre!

59
Cały nasz Świat tworzą procesy nieodwracalne …

60
Cały nasz Świat tworzą procesy nieodwracalne …

61
Cały nasz Świat tworzą procesy nieodwracalne …

62
Cały nasz Świat tworzą procesy nieodwracalne …

63
Cały nasz Świat tworzą procesy nieodwracalne …

64
Cały nasz Świat tworzą procesy nieodwracalne …

65
w warunkach izochorycznych możliwe tylko procesy egzotermiczne ≤ 0 ?
Dlaczego ???? A może „zasada minimalizacji energii”? dU ≤ 0 ? dU = -pzdV + dQ ≤ 0 ? dla V = const dQ ≤ 0 ? w warunkach izochorycznych możliwe tylko procesy egzotermiczne ≤ 0 ?

66
Eksperyment z kartami Jakie jest prawdopodobieństwo powrotu do pierwotnego, uporządkowanego rozkładu poprzez tasowanie ? Liczba wszystkich konfiguracji (kolejności kart) wynosi 52! Ω = 52! Jeśli wszystkie konfiguracje są jednakowo prawdopodobne, to prawdopodobieństwo zaistnienia jednej z nich wynosi
 wynosi 52! Ω = 52! Jeśli wszystkie konfiguracje są jednakowo prawdopodobne, to prawdopodobieństwo zaistnienia jednej z nich wynosi.")
67
II zasada termodynamiki - swobodna ekspansja gazu – przykład procesu nieodwracalnego
Początek - 1

68
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
2

69
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
3

70
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
4

71
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
5

72
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
6

73
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
7

74
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
8

75
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
8

76
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
7

77
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
6

78
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
5

79
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
4

80
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
3

81
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
2

82
II zasada termodynamiki - swobodna ekspansja gazu – przykład procesu nieodwracalnego
Początek - 1

83
Swobodna ekspansja gazu – przykład procesu nieodwracalnego
1 2 3 4 5 6 7 8
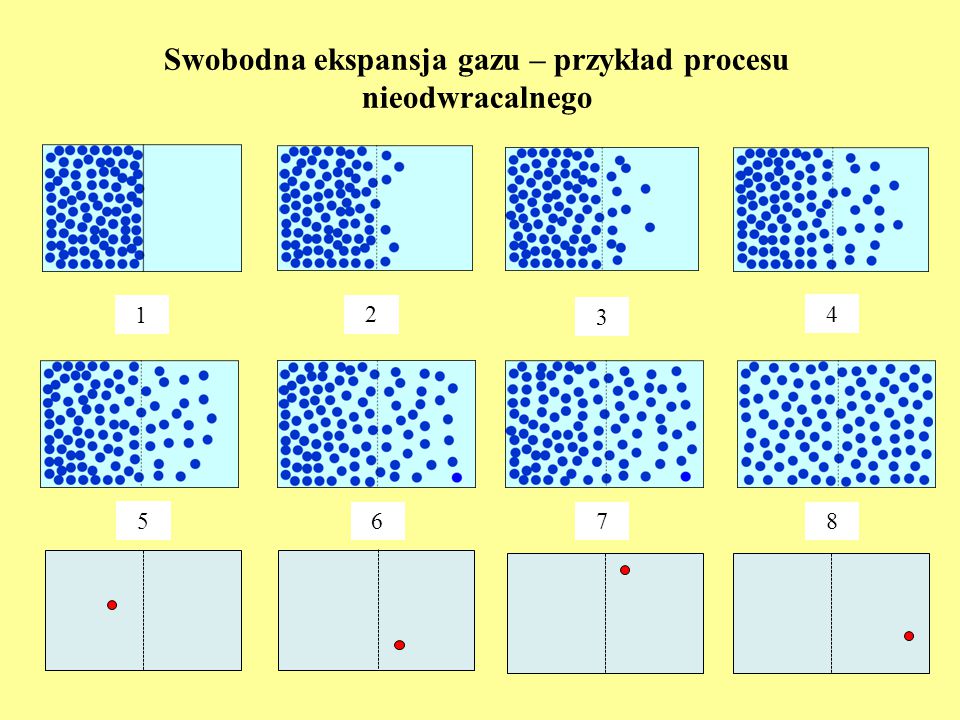
84
Swobodna ekspansja gazu
Każdej cząsteczce możemy przydzielić jeden z dwóch stanów – L i P. Cząsteczka w każdym z nich może się znaleźć z jednakowym prawdopodobieństwem. Liczba wszystkich możliwych stanów wynosi: 2∙2∙2∙2∙… = 2N. Przy założeniu, że wszystkie lokalizacje każdej cząsteczki są jednakowo prawdopodobne, prawdopodobieństwo powrotu do stanu początkowego wynosi Jeśli przyjąć, że zmiana konfiguracji odbywa się w czasie Plancka, tj. = s, przejście po wszystkich konfiguracjach wymagałoby czasu rzędu 2N lat! To znacznie dłużej niż istnieje (i będzie istniał) Wszechświat!
 Wszechświat!")
85
Entropia w ujęciu statystycznym
Jednemu stanowi makroskopowemu odpowiada olbrzymia liczba mikrostanów kwantowych Jeśli wszystkie stany są jednakowo osiągalne, to spontaniczny proces w układzie izolowanym biegnie od stanu 1 do stanu 2, jeśli Ω1 << 2 Wprowadzając funkcję zwaną entropią S = k lnΩ formułujemy zasadę wzrostu entropii: Dla każdego spontanicznego procesu zachodzącego w układzie izolowanym, tj. U, V, N = const, entropia musi rosnąć, osiągając maksimum w stanie równowagi

86
Stanowi makroskopowemu odpowiada wielka liczba mikrostanów kwantowych.
Podsumowanie Znaczenie funkcji S = k ln Stanowi makroskopowemu odpowiada wielka liczba mikrostanów kwantowych. Proces nieodwracalny przebiega od stanu mniej prawdopodobnego (realizowanego przez mniejszą liczbę mikrostanów kwantowych) do stanu bardziej prawdopodobnego (realizowanego przez większą liczbę mikrostanów kwantowych). Stanowi równowagi odpowiada maksymalna liczba mikrostanów kwantowych. Równoważne sformułowanie posługuje się pojęciem entropii. Odpowiednia reguła, zwana zasadą wzrostu entropii brzmi: Dla (N,V,U=const, tj. dla układu izolowanego) możliwy jest tylko proces, któremu towarzyszy wzrost entropii, która osiąga maksimum w stanie równowagi.
 do stanu bardziej prawdopodobnego (realizowanego przez większą liczbę mikrostanów kwantowych). Stanowi równowagi odpowiada maksymalna liczba mikrostanów kwantowych. Równoważne sformułowanie posługuje się pojęciem entropii. Odpowiednia reguła, zwana zasadą wzrostu entropii brzmi: Dla (N,V,U=const, tj. dla układu izolowanego) możliwy jest tylko proces, któremu towarzyszy wzrost entropii, która osiąga maksimum w stanie równowagi.")
87
Właściwości entropii S = kln Ω
Ponieważ dla układu złożonego, Ω = Ω1 ∙2 - entropia jest funkcją ekstensywną S = S1 + S2

88
Przyjmijmy, że parametrem niezależnym jest U1, wtedy
Znaczenie pochodnej izolacja od otoczenia N = const V = const U1 + U2 = const N1,V1 N2 ,V2 U1 U2 Jaki będzie warunek równowagi względem przepływu energii pomiędzy 1 a 2 ? Zgodnie z zasadą wzrostu entropii, stan równowagi odpowiada maksimum entropii S = S1 + S2 dU1 + dU2 = 0 dU1 = - dU2 Przyjmijmy, że parametrem niezależnym jest U1, wtedy

89
Definicja temperatury termodynamicznej
Znaczenie pochodnej izolacja od otoczenia N = const V = const U1 + U2 = const N1,V1 N2 ,V2 U1 U2 W stanie równowagi Definicja temperatury termodynamicznej

90
Związek pomiędzy termodynamiką statystyczną a klasyczną
stała Boltzmanna Jeśli przyjmiemy, że k = R/NA

91
dU = dw + dQ dU = dwodw + dQodw TdS = dQodw ≥ dQ Ciepło a entropia
praca odwracalna - dwodw jest minimalna ciepło odwracalne – dQodw jest maksymalne dQodw ≥ dQ TdS =

92
II zasada termodynamiki
Postuluje się istnienie funkcji stanu zwanej entropią (S), która ma następujące właściwości Jest funkcją ekstensywną
, która ma następujące właściwości. Jest funkcją ekstensywną.")
93
Lokalny charakter II Zasady
II Zasada nie ma charakteru uniwersalnego, stosuje się jedynie do układów: - makroskopowych, - w stanie równowagi, - ergodycznych. Z braku uniwersalności wynikają liczne nieporozumienia i błędne interpretacje (do dnia dzisiejszego !)
")
94
Rudolf Julius Emmanuel Clausius (1822-1888)
Ludwig Eduard Boltzmann ( )
")
95
Wnioski z I i II zasady (1)
dU = dw + dQ = dwodw + dQodw dU = -pdV + TdS Wnioski: Istnienie związków pomiędzy parametrami (funkcjami) stanu. Uzasadnienie zasady Duhema (dwa parametry opisują różniczkę zupełną). Interpretacja temperatury i możliwe dalsze rozwinięcie dU.
 stanu. Uzasadnienie zasady Duhema (dwa parametry opisują różniczkę zupełną). Interpretacja temperatury i możliwe dalsze rozwinięcie dU.")
96
Wnioski z I i II zasady(2)
parametr intensywny – siła uogólniona deformacja parametru ekstensywnego dU = -pdV + TdS To jest bilans energii: praca +ciepło ! ….bo mogą być inne formy przekazywania energii ! „zwykła” siła

97
Wnioski z I i II Zasady (3)
dla procesu odwracalnego dla każdego procesu U,V,(N) = const …. entropia rośnie i osiąga maksimum w stanie równowagi (zasada wzrostu entropii)
 = const. …. entropia rośnie i osiąga maksimum w stanie równowagi (zasada wzrostu entropii)")
98
Wnioski z I i II Zasady (4)
dla procesu odwracalnego dla każdego procesu S,V,(N) = const …. energia wewnętrzna maleje i osiąga minimum w stanie równowagi
 = const. …. energia wewnętrzna maleje. i osiąga minimum w stanie równowagi.")
99
Wnioski z I i II Zasady (5)
Nie tylko entropia decyduje o naszym Świecie…. Parametrem rozstrzygającym o kierunku zachodzenia procesów mogą być różne funkcje (zwane potencjałami termodynamicznymi). Entropia jest potencjałem termodynamicznym dla U,V, N = const, podczas gdy dla warunków S,V,N = const, takim potencjałem jest energia wewnętrzna. Z praktycznego punktu widzenia najlepszy byłby potencjał „rządzący” procesami w warunkach dających się łatwo kontrolować (stałe parametry p, V, T)
. Entropia jest potencjałem termodynamicznym dla U,V, N = const, podczas gdy dla warunków S,V,N = const, takim potencjałem jest energia wewnętrzna. Z praktycznego punktu widzenia najlepszy byłby potencjał „rządzący procesami w warunkach dających się łatwo kontrolować (stałe parametry p, V, T)")
100
Wnioski z I i II Zasady (6) – pozostałe potencjały
Entalpia: H = U + pV U = H - pV dla procesu odwracalnego dla każdego procesu S,p,(N) = const …. entalpia maleje i osiąga minimum w stanie równowagi
 = const. …. entalpia maleje. i osiąga minimum w stanie równowagi.")
101
Wnioski z I i II Zasady (7) – pozostałe potencjały
Energia swobodna: F = U - TS U = F + TS dla procesu odwracalnego dla każdego procesu T,V,(N) = const …. energia swobodna maleje i osiąga minimum w stanie równowagi
 = const. …. energia swobodna maleje. i osiąga minimum w stanie równowagi.")
102
Wnioski z I i II Zasady (8) – pozostałe potencjały
Entalpia swobodna: G = H – TS = U + pV - TS U = G – pV + TS dla procesu odwracalnego dla każdego procesu T,p,(N) = const …. entalpia swobodna maleje i osiąga minimum w stanie równowagi
 = const. …. entalpia swobodna maleje. i osiąga minimum w stanie równowagi.")
103
Entalpia swobodna – najważniejszy potencjał termodynamiczny
Entalpia swobodna (energia Gibbsa, funkcja Gibbsa) G = H – TS różniczka zupełna pochodne cząstkowe relacja Maxwella
 G = H – TS. różniczka zupełna. pochodne cząstkowe. relacja Maxwella.")
104
Potencjały termodynamiczne – pochodne i różniczki
różniczka zupełna pochodne cząstkowe relacje Maxwella Entropia dS = (1/T)dU + (p/T)dV (S/U)V = 1/T (S/V)U = p/T Energia wewnętrzna dU = TdS - pdV (U/S)V = T (U/V)S = -p (T/V)S = - (p/S)V Entalpia dH = TdS +Vdp (H/S)p= T (H/p)S = V (T/p)S = (V/S)p Energia swobodna dF = -SdT - pdV (F/T)V = -S (F/V)T= -p (S/V)T = (p/T)V Entalpia swobodna dG = -SdT +Vdp (G/T)p= -S (G/p)T = V (S/p)T = - (V/T)p
dU + (p/T)dV. (S/U)V = 1/T. (S/V)U = p/T. Energia wewnętrzna. dU = TdS - pdV. (U/S)V = T. (U/V)S = -p. (T/V)S = - (p/S)V. Entalpia. dH = TdS +Vdp. (H/S)p= T. (H/p)S = V. (T/p)S = (V/S)p. Energia swobodna. dF = -SdT - pdV. (F/T)V = -S. (F/V)T= -p. (S/V)T = (p/T)V. Entalpia swobodna. dG = -SdT +Vdp. (G/T)p= -S. (G/p)T = V. (S/p)T = - (V/T)p.")
105
Potencjały termodynamiczne
Parametry Warunek S (II zasada) U,V (dS)U,V ≥ 0 U (I zasada) S,V (dU)S,V ≤ 0 H = U + pV S, p (dH)S,p ≤ 0 F = U - TS T, V (dF)T,V ≤ 0 G = H - TS T, p (dG)T,p ≤ 0
 U,V. (dS)U,V ≥ 0. U (I zasada) S,V. (dU)S,V ≤ 0. H = U + pV. S, p. (dH)S,p ≤ 0. F = U - TS. T, V. (dF)T,V ≤ 0. G = H - TS. T, p. (dG)T,p ≤ 0.")
106
Wnioski z I i II Zasady Termodynamiki
? 1. Istnieją funkcje (potencjały termodynamiczne), których zmiana, przy stałości dwóch parametrów, decyduje o kierunku procesu; potencjał termodynamiczny osiąga minimum (maksimum) w stanie równowagi. 2. Daje to możliwość znajdywania związków między parametrami w stanie równowagi. 3. Można wyprowadzić liczne tożsamości, wyrażające związki pomiędzy funkcjami termodynamicznymi, umożliwiające obliczanie ich zmian podczas rzeczywistych procesów.
, których zmiana, przy stałości dwóch parametrów, decyduje o kierunku procesu; potencjał termodynamiczny osiąga minimum (maksimum) w stanie równowagi. 2. Daje to możliwość znajdywania związków między parametrami w stanie równowagi. 3. Można wyprowadzić liczne tożsamości, wyrażające związki pomiędzy funkcjami termodynamicznymi, umożliwiające obliczanie ich zmian podczas rzeczywistych procesów.")
107
Zależność entropii od temperatury

108
Jak wyznaczyć entropię?
cp lnT lnT0 lnT1

109
III Zasada Termodynamiki
Jeśli przyjmiemy, że S(T=0) = 0 - postulat ten nosi nazwę III Zasady Termodynamiki, W termodynamice statystycznej wymóg ten jest zbyteczny, bo dla S(Ω =1) = kln(1) = 0 i ten stan odpowiada T = 0
 = 0. - postulat ten nosi nazwę III Zasady Termodynamiki, W termodynamice statystycznej wymóg ten jest zbyteczny, bo dla. S(Ω =1) = kln(1) = 0. i ten stan odpowiada T = 0.")
Podobne prezentacje











![ENTALPIA - H [ J ], [ J mol -1 ] TERMODYNAMICZNA FUNKCJA STANU dH = H 2 – H 1, H = H 2 – H 1 Mgr Beata Mycek - Zakład Farmakokinetyki i Farmacji Fizycznej.](/1/57659/big_thumb.jpg "ENTALPIA - H [ J ], [ J mol -1 ] TERMODYNAMICZNA FUNKCJA STANU dH = H 2 – H 1, H = H 2 – H 1 Mgr Beata Mycek - Zakład Farmakokinetyki i Farmacji Fizycznej.>")







>")