Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Część druga

2
Konduktometria Metoda elektroanalityczna oparta na pomiarze przewodnictwa elektrolitycznego, którego wartość zmienia się wraz ze zmianą stężenia jonów w analizowanym roztworze. Przewodnictwo elektrolityczne

3
Przewodnictwo elektrolityczne zależy od:

4
Przepływ prądu przez roztwory elektrolitów podlega prawu Ohma wg. którego natężenie prądu jest wprost proporcjonalne do przyłożonego napięcia U, a odwrotnie proporcjonalne do oporu R, jaki roztwór stawia prądowi. Opór przewodnika R zależy od jego długości l, przekroju s i rodzaju materiału, z którego wykonano przewodnik: gdzie: , [ cm], jest tzw. oporem właściwym l – oznacza odległość między elektrodami s – powierzchnię elektrod Przewodnictwo jest odwrotnością oporności elektrycznej roztworu pomiędzy dwiema obojętnymi elektrodami.

5
Dla elektrolitów zwykle podaje się przewodnictwo elektryczne (konduktancję), G: Jednostką przewodnictwa jest simens: S = -1 Po przekształceniu wzoru na przewodnictwo właściwe otrzymujemy: Stosunek, cm -1, oznacza się symbolem k i nazywa stałą naczynka elektrolitycznego lub pojemnością oporową naczynka.
, G: Jednostką przewodnictwa jest simens: S = -1 Po przekształceniu wzoru na przewodnictwo właściwe otrzymujemy: Stosunek, cm -1, oznacza się symbolem k i nazywa stałą naczynka elektrolitycznego lub pojemnością oporową naczynka.")
6
Przewodnictwo roztworu zawierającego 1 gramorównoważnik jonów pomiędzy elektrodami o powierzchni s (cm 2 ) odległymi od siebie o 1 cm, a więc zamykającymi objętość V, 1 litra roztworu nazywamy przewodnictwem równoważnikowym x albo x. gdzie: N oznacza liczbę gramorównoważników jonów w 1 litrze roztworu. Jeden gramorównoważnik to ilość jonów, która niesie ładunek jednego Faradaya F = 96500 kulombów (C).
..")
7
Przewodnictwem molowym nazywamy przewodnictwo elektryczne, jakie wykazuje warstwa elektrolitu w przestrzeni między dwiema elektrodami oddalonymi od siebie o 1 cm i o takiej objętości V, aby znajdował się w niej 1 mol substancji.,. Wymiarem przewodnictwa molowego 0 jest S · m 2 ∙ mol -1. Graniczna wartość do jakiej dąży przewodnictwo molowe, gdy stężenie elektrolitu dąży do zera, nazywa się granicznym przewodnictwem molowym.

8
Prawo Kohlrauscha o niezależnej wędrówce jonów, graniczne przewodnictwo molowe danego elektrolitu jest równe sumie granicznych przewodnictw jonowych (kationu i anionu w rozcieńczeniu nieskończenie wielkim).
.")
9
Ruchliwość (elektryczna) jonu u szybkość z jaką jon porusza się pod wpływem pola elektrycznego o jednostkowym natężeniu, V cm -1. Pomiędzy ruchliwością jonów w roztworze nieskończenie rozcieńczonym a granicznym przewodnictwem molowym zachodzi następująca zależność:, gdzie: F – oznacza stałą Faradaya, 96500 C

10
Molowe przewodnictwa jonowe nie zależą od stężenia. Arrhenius wyraził przewodnictwo molowe w skończonych stężeniach za pomocą następującej zależności uwzględniającej stopień dysocjacji . Stopień dysocjacji można zatem obliczyć jako stosunek przewodnictwa równoważnikowego w danym stężeniu do przewodnictwa równoważnikowego w rozcieńczeniu nieskończenie dużym:

11
Przewodnictwo roztworów rozcienczonych

12
Przewodnictwo roztworów stężonych

13
Wpływ temperatury na przewodnictwo właściwe NaCl

16
PotencjometriaPehametria

17
Potencjometria jest metodą wykorzystującą zależność między aktywnością oznaczanego jonu w roztworze, a potencjałem elektrycznym elektrody. Praktycznie wyznacza się stężenie oznaczanego składnika na podstawie siły elektromotorycznej SEM ogniwa utworzonego z elektrody wskaźnikowej i porównawczej (dla roztworów rozcieńczonych). Zależność tę opisuje najogólniej równanie Nernsta
. Zależność tę opisuje najogólniej równanie Nernsta.")
18
E 0 - normalny potencjał elektrody (a M n+ = 1) R - stała gazowa (8,31441 J K -1 mol -1 ) T - temperatura bezwzględna (K) F - stała Faraday’a (96486,7± 0,54 C mol -1 ) n - liczba elektronów biorąca udział w reakcji a M n+ - aktywność jonów metalu a M 0 = 1
 R - stała gazowa (8,31441 J K -1 mol -1 ) T - temperatura bezwzględna (K) F - stała Faraday’a (96486,7± 0,54 C mol -1 ) n - liczba elektronów biorąca udział w reakcji a M n+ - aktywność jonów metalu a M 0 = 1")
19
Elektrody wskaźnikowe pierwszego rodzaju Elektrody wskaźnikowe drugiego rodzaju Elektrody wskaźnikowe trzeciego rodzaju Elektrody wskaźnikowe utleniająco-redukujące Elektrody porównawcze Elektrody wskaźnikowe jonoselektywne

20
Właściwości: * Odwracalne względem kationu * Zbudowane z metalu (lub gazu) zanurzonego w roztworze własnych jonów * Reakcja przebiegająca na elektrodzie M o M n+ + n e - * Ważną elektrodą pierwszego rodzaju jest elektroda wodorowa ½ H 2(g) H + + e -
 zanurzonego w roztworze własnych jonów * Reakcja przebiegająca na elektrodzie M o M n+ + n e - * Ważną elektrodą pierwszego rodzaju jest elektroda wodorowa ½ H 2(g) H + + e -")
21
Właściwości: * Odwracalne względem wspólnego anionu * Składają się z metalu pokrytego trudno rozpuszczalną solą tego metalu, zanurzonego w roztworze zawierającym ten anion * Reakcja zachodząca na elektrodzie: M 0 + A - MA + e - * Ważne elektrody drugiego rodzaju: chlorosrebrowa, kalomelowa

22
Właściwości * Odwracalne względem wspólnego kationu * Tworzą je metale otoczone cienką warstwą trudno rozpuszczalnej soli tego metalu oraz warstwą soli nieco lepiej rozpuszczalnej, zawierającej ten sam anion * Reakcja elektrodowa zachodzi według równania: M 0 + M (b) A M (a) A + M + (b) + e - * Przykład: Pb|PbC 2 O 4 ||CaC 2 O 4 |Ca 2+ * Obecnie rzadko stosowane
 A M (a) A + M + (b) + e - * Przykład: Pb|PbC 2 O 4 ||CaC 2 O 4 |Ca 2+ * Obecnie rzadko stosowane")
23
Budowa elektrod utleniająco-redukcyjnej * Obojętny chemicznie metal (Pt,Au) zanurzony w roztworze zawierającym substancje w postaci zarówno utlenionej jak i zredukowanej * Przykład: elektroda chinhydronowa
 zanurzony w roztworze zawierającym substancje w postaci zarówno utlenionej jak i zredukowanej * Przykład: elektroda chinhydronowa")
24
Cechy idealnej elektrody porównawczej: * Stałość potencjału * Odtwarzalność potencjału i brak histerezy temperaturowej * Uniwersalność zastosowań i prostota obsługi * Mały opór elektryczny * Odtwarzalny i niski potencjał dyfuzyjny * Mały wypływ elektrolitu elektrody do badanego roztworu Najczęściej elektrodami odniesienia są elektrody drugiego rodzaju: elektroda chlorosrebrowa i nasycona elektroda kalomelowa

25
Cechy membranowych elektrod jonoselektywnych (ISE): * Elektrodowo czynną częścią elektrody jest membrana * O różnicy potencjału na granicy faz membrana/roztwór decyduje reakcja wymiany jonowej między jonami z roztworu a jonami membrany.
: * Elektrodowo czynną częścią elektrody jest membrana * O różnicy potencjału na granicy faz membrana/roztwór decyduje reakcja wymiany jonowej między jonami z roztworu a jonami membrany.")
26
Elektrody jonoselektywne można podzielić ze względu na stan skupienia fazy tworzącej membranę * Elektrody szklane * Elektrody ze stałymi membranami * Elektrody z membranami ciekłymi * Elektrody z podwójnymi membranami (czułe na gazy, enzymatyczne)
")
27
Zakres prostoliniowości wskazań * Nazywany zakresem odpowiedzi nernstowskiej elektrody * Szeroki zakres prostoliniowości zapewnia dobre pomiary w bardzo dużym zakresie stężeń czułość * Miarą czułości jest nachylenie krzywej kalibracyjnej * Selektywność * Miarą selektywności jest współczynnik selektywności K w równaniu Nikolskiego. Wartość liczbowa współczynnika oznacza ile razy silniej elektroda reaguje zmianą potencjału na obecność jonu głównego w porównaniu z jonem zakłócającym

29
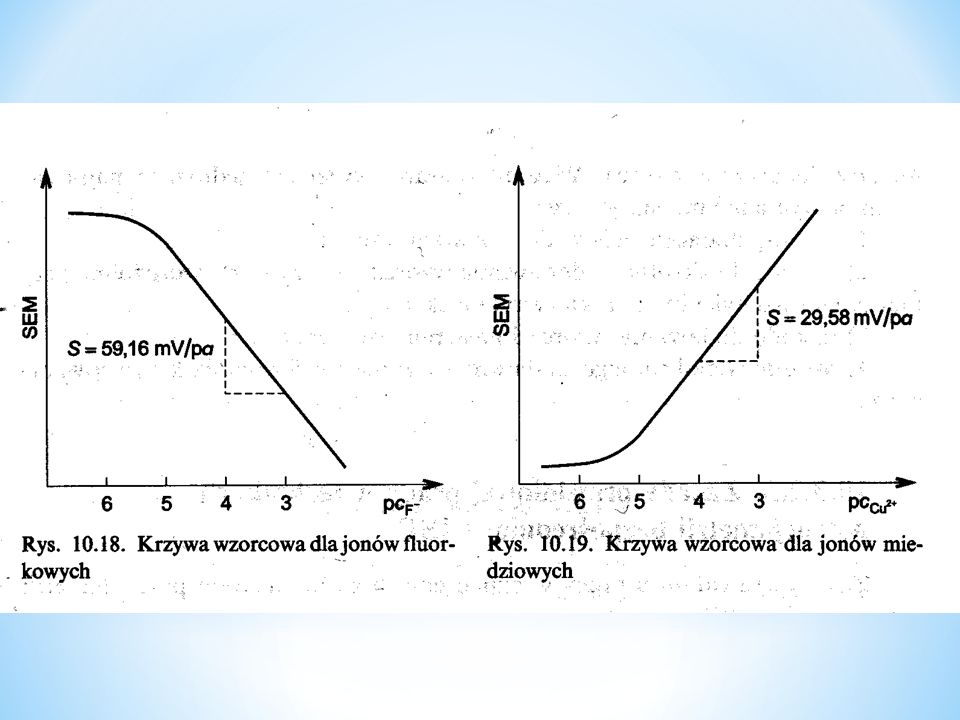
Elektrody szklane są pierwszymi elektrodami membranowymi wykorzystywanymi w analizie potencjometrycznej (Haber, Klemensiewicz, 1909). Są one powszechnie stosowane do oznaczeń pH. Elektroda szklana składa się z rurki szklanej zakończonej cienkościenna banieczką. Wewnątrz znajduje się elektroda chlorosrebrowa zanurzona w roztworze wewnętrznym HCl o stałej aktywności jonów lub w buforze pH. W handlu dostępne są elektrody kombinowane zawierające dodatkowo elektrodę odniesienia. Elektrody szklane nie wymagają dodatkowych odczynników, potencjał ustala się szybko, zakres pH możliwy do oznaczenia jest bardzo szeroki, od pH 1 do 12.

30
Potencjometria jako metoda elektroanalityczna jest metodą analizy ilościowej roztworów. Można wyróżnić metody: * pH-metria * Miareczkowanie potencjometryczne * Potencjometria bezpośrednia (pH-metria stanowi oddzielna, wyspecjalizowaną technikę potencjometryczną) * Analiza kliniczna: poziom glukozy, stężenie elektrolitów we krwi * Działania policji -alkomaty
 * Analiza kliniczna: poziom glukozy, stężenie elektrolitów we krwi * Działania policji -alkomaty.")
31
Mierząc stężenie jonów wodorowych wkraczamy w zakres pehametrii. pH jest ujemnym log [H + ] z wartości stężenia jonów wodorowych. Pomiaru stężenia jonów dokonuje się mierząc SEM ogniwa złożonego z dwóch elektrod: elektrody pracującej i elektrody odniesienia (wchodzących w skład tzw. elektrody kombinowanej)
.")
32
1 - elektroda wyprowadzająca chlorosrebrowa 2 - roztwór wewnętrzny elektrody chlorosrebrowej (0,1 mol/dm 3 HCl) 3 - membrana szklana (niskooporowa;100-500 MW) 4 - elektroda porównawcza chlorosrebrowa 5 - roztwór wewnętrzny elektrody porównawczej (nasycony roztwór KCl nasycony AgCl) 6 - przewód wyprowadzający sygnał 7 - wlew roztworu wewnętrznego
 3 - membrana szklana (niskooporowa; MW) 4 - elektroda porównawcza chlorosrebrowa 5 - roztwór wewnętrzny elektrody porównawczej (nasycony roztwór KCl nasycony AgCl) 6 - przewód wyprowadzający sygnał 7 - wlew roztworu wewnętrznego")
35
* Metoda krzywych wzorcowych –polega na wykreśleniu krzywej wzorcowej dzięki której można wyznaczyć nieznaną aktywność oznaczanego jonu. Możliwa do zastosowania przy szerokim prostoliniowym zakresie. * Metoda dodatku wzorca -polega na przeprowadzeniu pomiarów potencjału przed i po dodaniu ilościowo odmierzonej ilości roztworu wzorcowego

36
Za pomocą pomiarów potencjometrycznych można śledzić przebieg miareczkowania. Ważny jest odpowiedni dobór elektrody wskaźnikowej Punkt końcowy miareczkowania można wyznaczyć * Metodą graficzną * Metodą pierwszej pochodnej * Metodą drugiej pochodnej * Metodami numerycznymi

37
Biologiczne układy redoks

38
Fluorescencja i metody fluorescencyjne. Polaryzacja fluorescencji. Gaszenie fluorescencji

39
Fluorescencja – emisja promieniowania przez niektóre cząsteczki o innej długości fali niż światło uprzednio pochłonięte. Proces ten stanowi podstawę spektrofluorymetrii - techniki pomiarowej, której wysoka czułość jest wykorzystywana dla śledzenia subtelnych zmian stężenia i właściwości makrocząsteczek biologicznych.

40
Luminescencja Emisja fotonów (w zakresie ultrafioletu, widzialnym i podczerwonym) z elektronowych stanów wzbudzonych. Fotoluminescencja Elektroluminescencja Chemiluminescencja Tryboluminescencja Termoluminescencja Bioluminescencja Sonoluminescencja

41
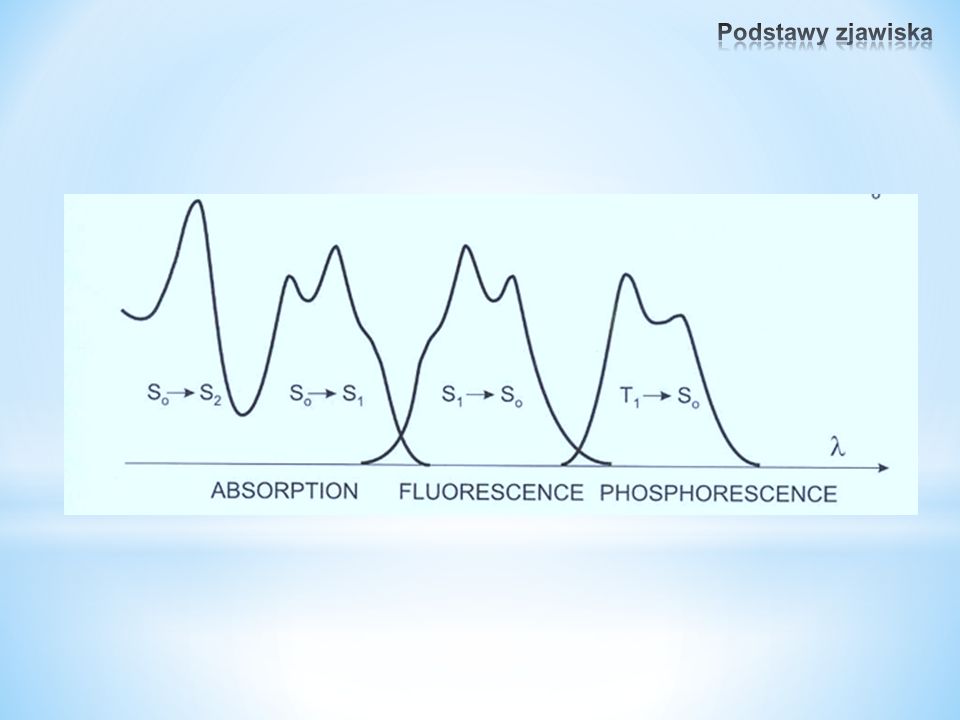
Fotoluminescencja Fluorescencja opóźniona Termiczna (T 1 → S 1, mała różnica energii, czas życia T 1 długi) Zderzenia T 1 + T 1 energia na powrót do S 1 Fluorescencja Ze stanów singletowych Fosforescencja Ze stanów trypletowych
 Zderzenia T 1 + T 1 energia na powrót do S 1 Fluorescencja Ze stanów singletowych Fosforescencja Ze stanów trypletowych")
42
http://micro.magnet.fsu.edu/primer/java/jablonski/lightandcolor/ Czas

44
Przejście międzysystemowe (interkombinacyjne), ISC Konwersja wewnętrzna, IC hv Cząsteczka wzbudzona fluorescencjaZmiany konformacyjne Transfer elektronu Przekształcenia fotochemiczne Ekscymery i ekscypleksy Transfer energii fosforescencja Fluorescencja opóźniona
, ISC Konwersja wewnętrzna, IC hv Cząsteczka wzbudzona fluorescencjaZmiany konformacyjne Transfer elektronu Przekształcenia fotochemiczne Ekscymery i ekscypleksy Transfer energii fosforescencja Fluorescencja opóźniona")
45
Absorpcja10 -15 s Relaksacja oscylacyjna10 -12 - 10 -10 s Czas życia stanu S 1 10 -10 - 10 -17 s Przejście międzysystemowe10 -10 - 10 -8 s Wewnętrzna konwersja10 -11 - 10 -9 s Czas życia stanu T 1 10 -6 - 1 s

46
Widmo Maksimum emisji λ em Szerokość pasm Ilość pasm Wydajno ść kwantowa Φ = fotony wyemitowane/fotony zaabsorbowane Czas ż ycia fluorescencji Opóźnienie pomiędzy absorpcją a emisją

47
Różnica energii pomiędzy położeniem maksimum na widmie absorpcji a położeniem maksimum na widmie emisji (wyrażona w częstościach lub długościach fali) https://lp.uni-goettingen.de/get/image/6754
")
48
Powstawanie pasm anty-stockesowskich

49
* Duża molowa absorpcja w rejonie wzbudzenia * Wysoka wydajność kwantowa * Fotostabilność * Długi czas życia w stanie wzbudzonym * Duże przesunięcie Stokesa

50
Efekt ciężkiego atomu Związki karbonylowe SO 3 H NH 2, OH etc. Zmiana przejścia o najniższej energii z π →π* na n→π* Dominuje przejście międzysystemowe (ISC)
.")
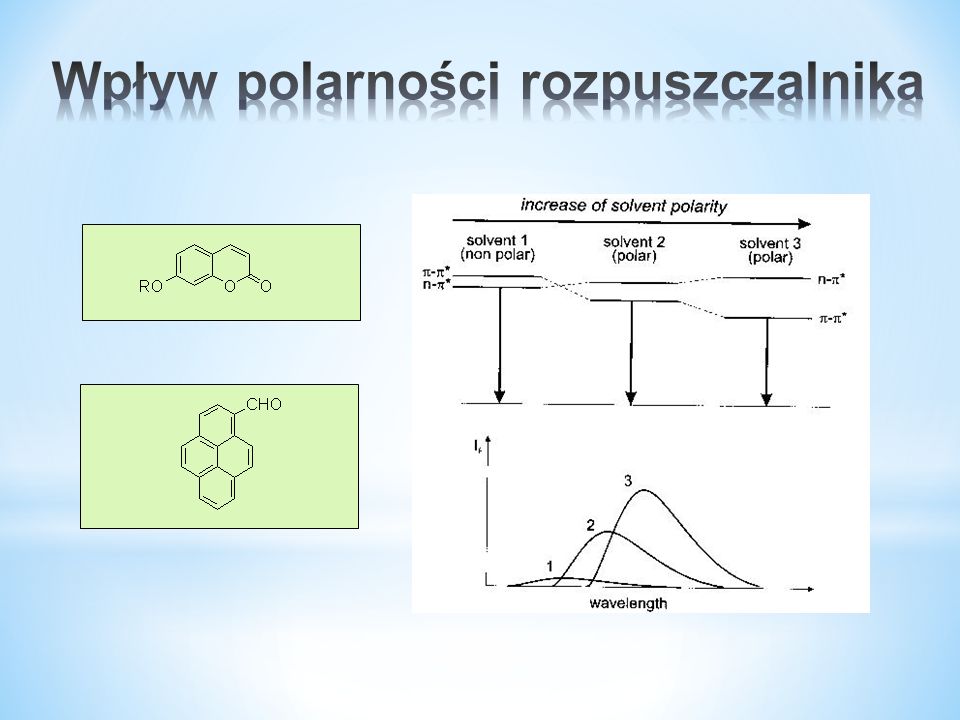
51
n→π* ma niższą energię niż π →π*, ale gdy są wiązania wodorowe może to ulec odwróceniu. Tak więc Φ wzrasta ze wzrostem H-donorowości rozpuszczalnika.

52
Usztywnienie cząsteczki zmniejsza możliwości przejść bezpromienistych a tym samym prowadzi do wyższej wydajności kwantowej fluorescencji. Φ = 0.54 Φ = 0.91

54
* Sensory * Wizualizacja związków biologicznie czynnych w komórkach * Mikroskopia fluorescencyjna * Polarność rozpuszczalnika * Pomiary gęstości cieczy

55
DziedzinaInformacje Błony biologiczneOddziaływania białko-lipidy, potencjał mitochondrialny, lokalizacja białek, efekty dodatków BiałkaDenaturacja, dynamika, przemiany konformacyjne Kwasy nukleinoweDynamika, str. helikalna, deformacje (też fotofizyczne), dostępność KomórkiWizualizacja membran, DNA, RNA, aktywność enzymów, H +, Na +, K +, transport przez błony PolimeryDynamika, rozdział faz, dyfuzja Roztwory surfaktantów Krytyczne stężenia micelli, przemiany fazowe
, dostępność KomórkiWizualizacja membran, DNA, RNA, aktywność enzymów, H +, Na +, K +, transport przez błony PolimeryDynamika, rozdział faz, dyfuzja Roztwory surfaktantów Krytyczne stężenia micelli, przemiany fazowe.")
57
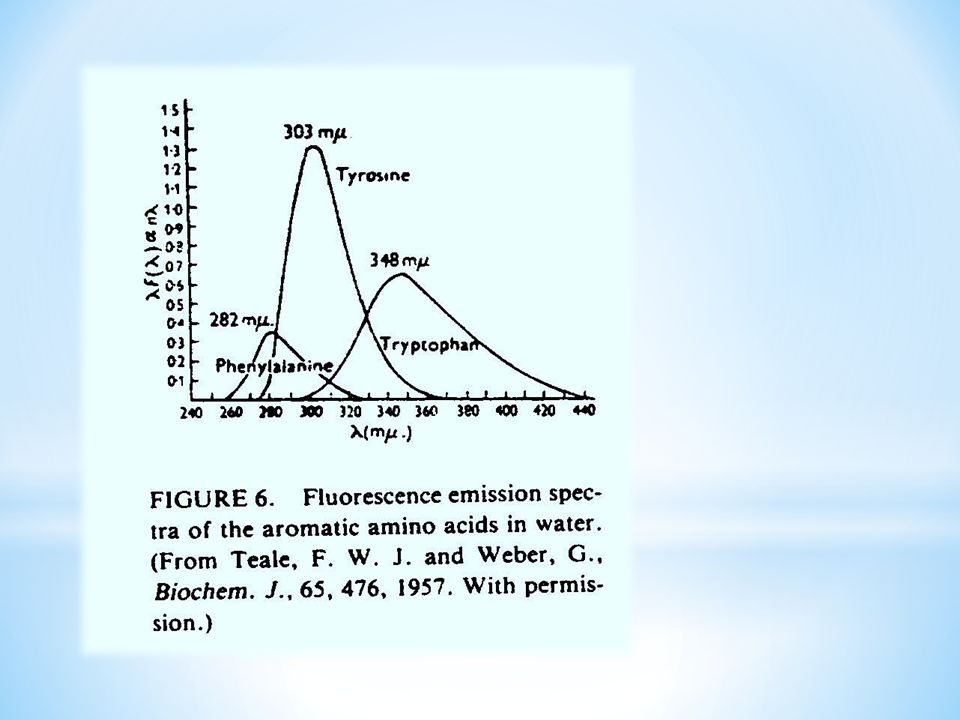
FLUORESCENCJA ZWIAZKÓW BIOLOGICZNIE CZYNNYCH. Substancjami wykazującymi zjawisko fluorescencji wśród związków biologicznie czynnych są miedzy innymi: Aminokwasy aromatyczne: tryptofan, tyrozyna, fenyloalanina Zasady nukleinowe w DNA i RNA: adenina, guanina, cytozyna, tymina, uracyl Barwniki roślinne: chlorofile, bakteriochlorofile i karotenoidy Witaminy i hormony: np. ryboflawina (wit. B2)
.")
58
Fluorescencja związków: Wewnętrzna Np. tryptofan, zawierający w swojej strukturze grupę Indolową (długość fali wzbudzenia 280-295 nm/długość fali emisji 320-350 nm, zależnie od polarności otoczenia tryptofanu, efekt solwatochromowy) Białka zawierające w cząsteczce aminokwasy aromatyczne Zewnętrzna gdy badany związek nie wykazuje fluorescencji, możliwa jest jego modyfikacja chemiczna za pomocą fluoroforu zewnętrznego. Jednym z przykładów jest izotiocyjanian fluoresceiny (FITC), który reaguje z wolnymi grupami aminowymi obecnymi w białkach.
 Białka zawierające w cząsteczce aminokwasy aromatyczne Zewnętrzna gdy badany związek nie wykazuje fluorescencji, możliwa jest jego modyfikacja chemiczna za pomocą fluoroforu zewnętrznego. Jednym z przykładów jest izotiocyjanian fluoresceiny (FITC), który reaguje z wolnymi grupami aminowymi obecnymi w białkach..")
59
Wskaźniki/znaczniki fluorescencyjne Grupa wskaźników chemicznych stosowanych w biologii molekularnej i analityce medycznej. W punkcie końcowym miareczkowania następuje zmiana fluorescencji próbki (np. fluoresceina, kumaryna); W biologii komórki - barwienie komórek. Przykładami mogą być stosowane do barwienia komórkowego jodek propidyny, Hoechst, oranż akrydyny, kalceina, błękit trypanu.
; W biologii komórki - barwienie komórek. Przykładami mogą być stosowane do barwienia komórkowego jodek propidyny, Hoechst, oranż akrydyny, kalceina, błękit trypanu..")
60
Izotiocyjanian fluoresceiny, FITC Błękit trypanu Jodek propidyny

61
Kalceina Kalceina, AM

63
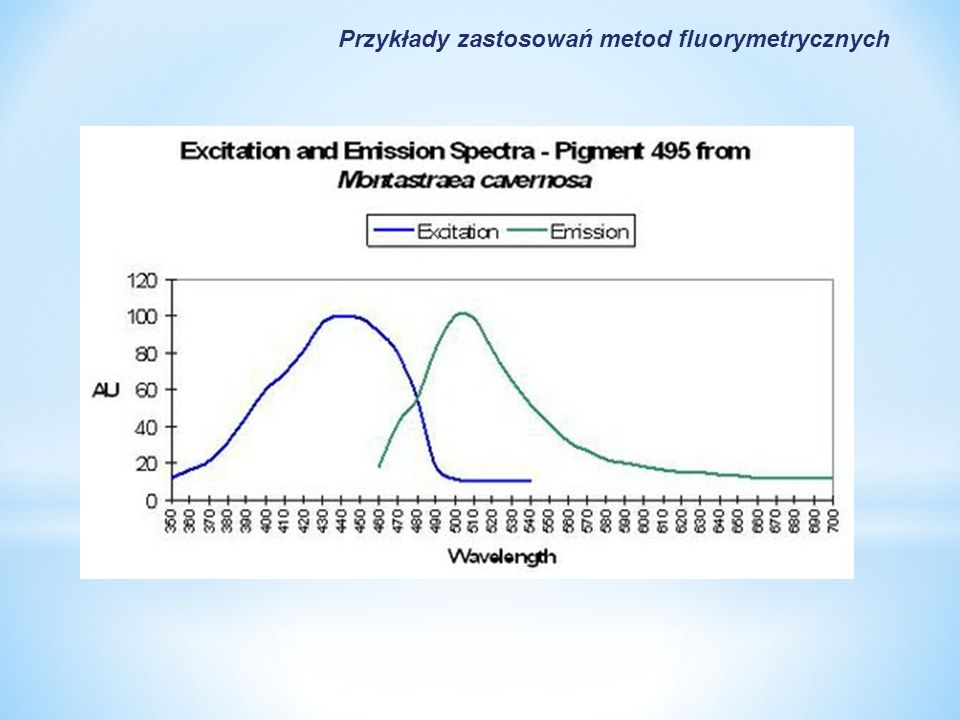
Biologia molekularna: 1.Fluorescencja tryptofanowa jest szeroko stosowana do badania i monitorowania zmian konformacyjnych białek spowodowanych lokalnymi zmianami struktury i dynamiki białek; 2. Znaczniki fluorescencyjne są wskaźnikami śmierci komórkowej, jej rodzaju i etapu; 3.Badanie zmian potencjału błonowego Monitoring środowiska: 1.Oznaczanie skuteczności pasteryzacji mleka poprzez fluorymetryczne oznaczanie aktywności fosfatazy alkalicznej (ALP). Metodą referencyjną jest metoda fluorymetryczna według PN-EN ISO 11816; 2.Oznaczanie jakości koralowców; 3.Wykrywanie zafałszowań oliwy ekstra virgin olejami z nasion (sojowymi, słonecznikowymi i rzepakowymi) metodą synchronicznych widm fluorescencji. Przykłady zastosowań metod fluorymetrycznych Oznaczenia jakościowe
. Metodą referencyjną jest metoda fluorymetryczna według PN-EN ISO 11816; 2.Oznaczanie jakości koralowców; 3.Wykrywanie zafałszowań oliwy ekstra virgin olejami z nasion (sojowymi, słonecznikowymi i rzepakowymi) metodą synchronicznych widm fluorescencji. Przykłady zastosowań metod fluorymetrycznych Oznaczenia jakościowe.")
64
Przykłady zastosowań metod fluorymetrycznych Oznaczenia ilościowe Oznaczanie stężeń substancji fluoryzujących. W pewnym zakresie stężeń fluorescencja próbki jest wprost proporcjonalna do stężenia substancji oznaczanej (analitu).
..")
65
Statyczne Dynamiczne Stężeniowe Chemiczne Gaszenie fluorescencji

66
Komórki barwione kalceiną i jodkiem propidyny Przykłady zastosowań metod fluorymetrycznych Komórki barwione Hoechst 33342 i jodkiem propidyny

67
Jodek propidyny (PI) – widma fluorescencji i ekscytacji Kalceina – widma fluorescencji i ekscytacji Przykłady zastosowań metod fluorymetrycznych
 – widma fluorescencji i ekscytacji Kalceina – widma fluorescencji i ekscytacji Przykłady zastosowań metod fluorymetrycznych")
69
Gaszenie fluorescencji

71
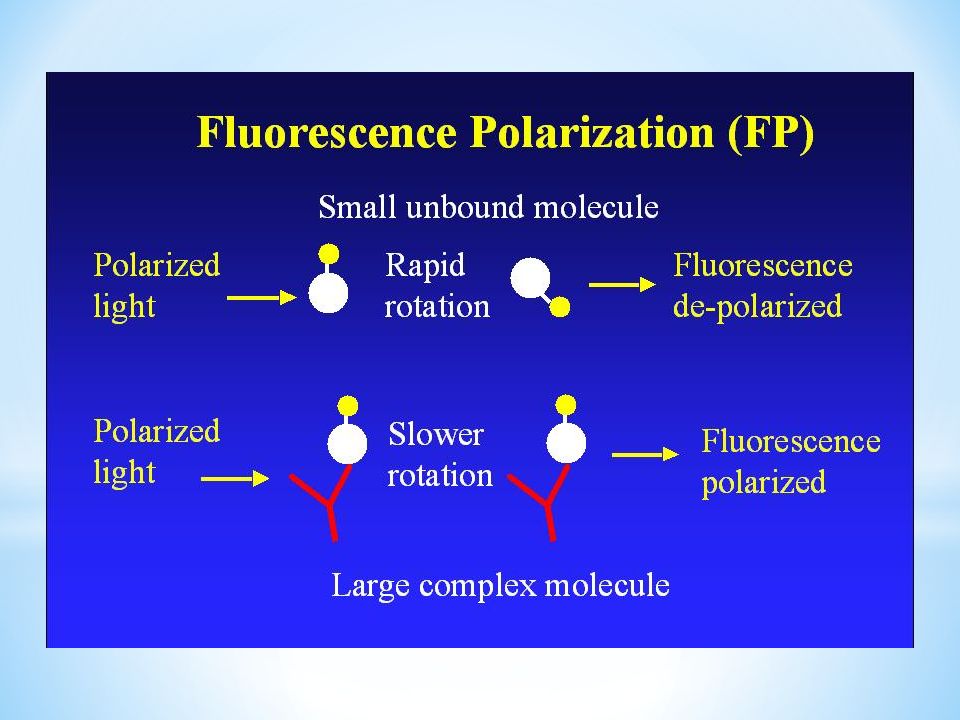
Polaryzacja fluorescencji

72
Schemat spektrofluorymetru klasycznego

73
https://i.publiclab.org
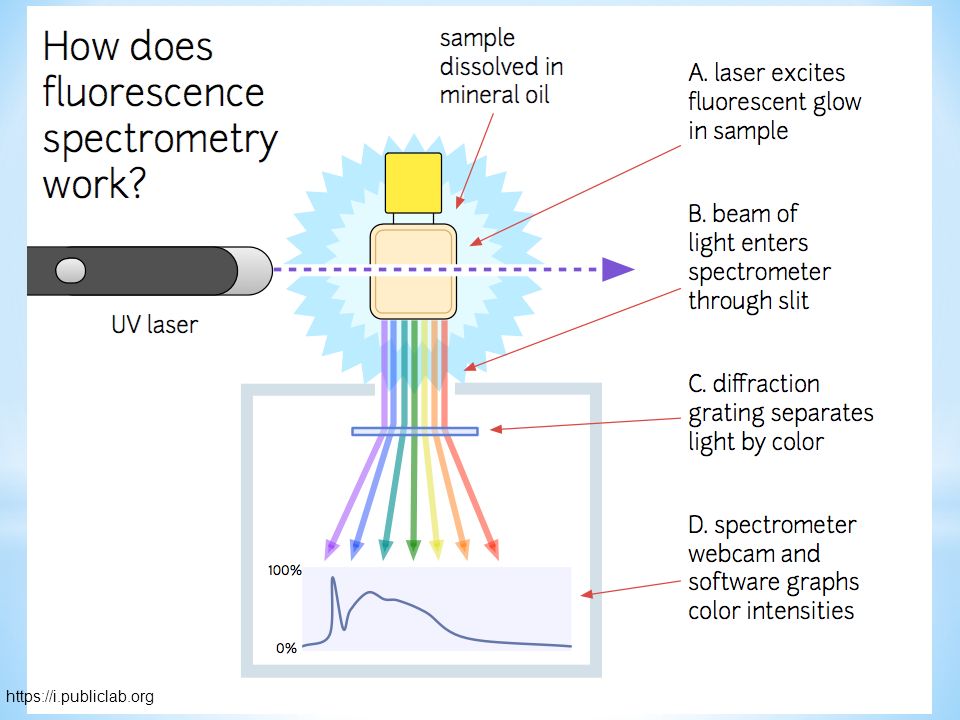
74
Spektrofluorymetr

75
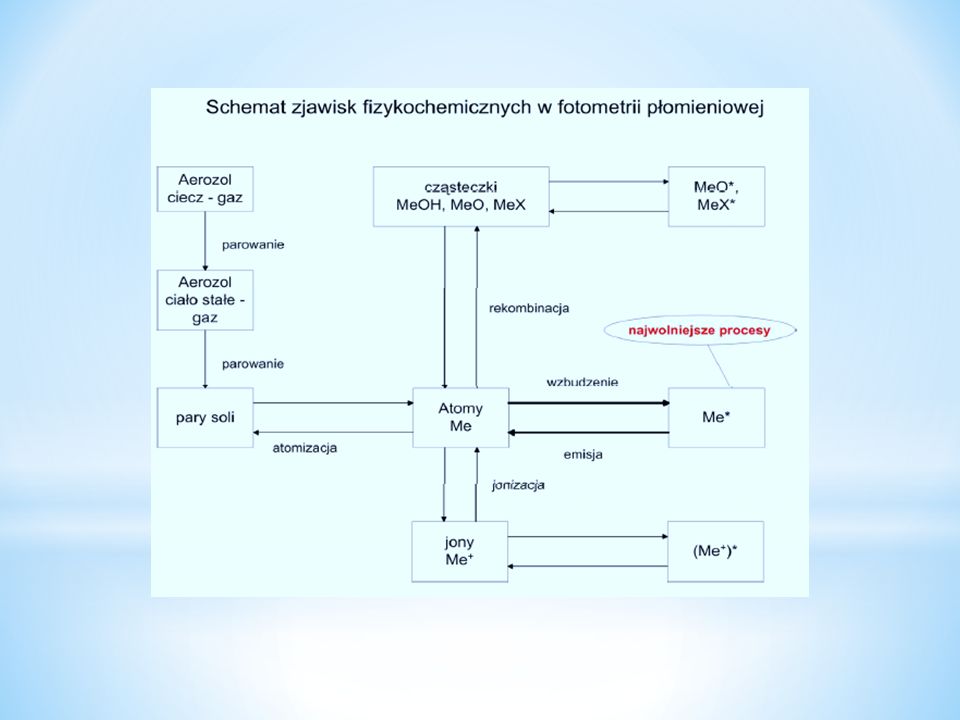
Fotometria płomieniowa. Absorpcyjna spektrometria atomowa

77
Linie widmowe różnych atomów

78
Widmo emisyjne atomu żelaza

84
Schemat blokowy fotometru płomieniowego

86
Rodzaje fotometrów płomieniowych

87
Wykonywanie pomiarów

89
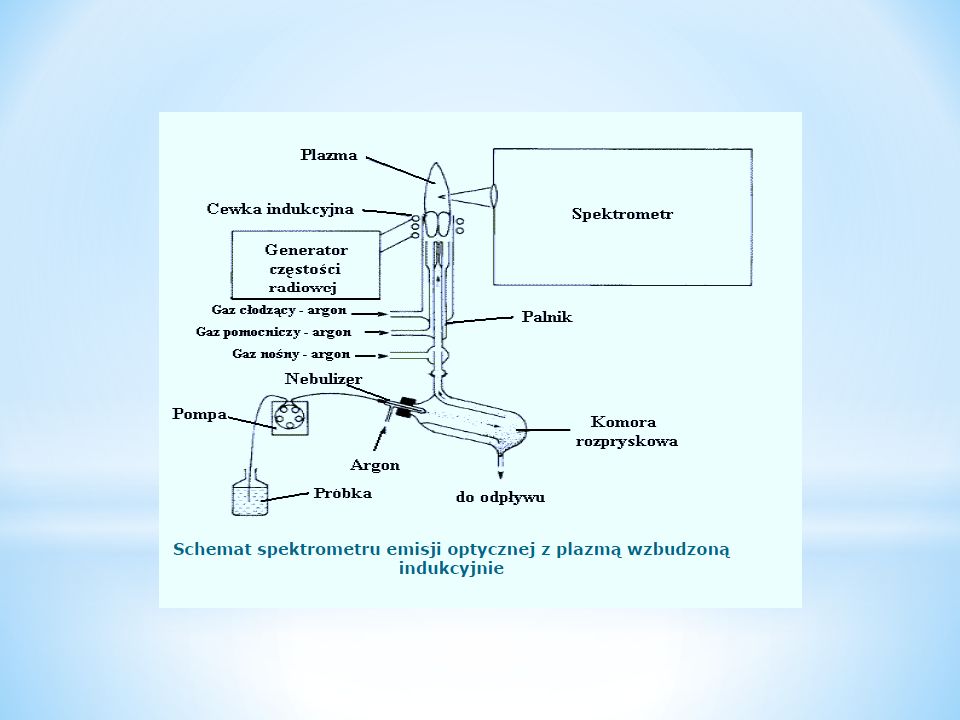
Plazma istnieje w temp. powyżej 6000 o C. Jest to gaz, którego atomy lub cząsteczki w większym lub mniejszym stopniu rozpadają się na dodatnio naładowane nośniki. Temperatura - 10000 K w przypadku generatorów wysokiej częstotliwości INDUKCYJNIE WZBUDZANA PLAZMA – ICP-AES (Inductively Coupled Plasma – Atomic Emission Spectroscopy) Zalety: - brak absorpcji wewnątrz źródła - brak odwrócenia linii w strefie niskotemperaturowej - brak zakłóceń związanych z obecnością tlenu (np. w przypadku Si lub B) - możliwość oznaczania trudnowzbudzalnych atomów (Cl, Br, I, S)
 Zalety: - brak absorpcji wewnątrz źródła - brak odwrócenia linii w strefie niskotemperaturowej - brak zakłóceń związanych z obecnością tlenu (np. w przypadku Si lub B) - możliwość oznaczania trudnowzbudzalnych atomów (Cl, Br, I, S).")
90
W metodzie ICP źródłem wzbudzenia jest plazma argonowa (lub hel), wytwarzana w palniku w następujący sposób: gazowy argon (99,995 %), jest wprowadzany do rury kwarcowej, owiniętej w górnej części cewką indukcyjną połączoną z generatorem częstości radiowej; najczęściej stosowane częstości radiowe to 27 lub 41 MHz; energia jonizacji 4 - 26 eV ICP-OES (lub ICP-AES) - pozwala oznaczyć jednocześnie około 70 pierwiastków w różnorodnych matrycach.
, wytwarzana w palniku w następujący sposób: gazowy argon (99,995 %), jest wprowadzany do rury kwarcowej, owiniętej w górnej części cewką indukcyjną połączoną z generatorem częstości radiowej; najczęściej stosowane częstości radiowe to 27 lub 41 MHz; energia jonizacji eV ICP-OES (lub ICP-AES) - pozwala oznaczyć jednocześnie około 70 pierwiastków w różnorodnych matrycach.")
93
Zalety techniki ICP – OES umożliwia analizę zarówno jednego pierwiastka jak i analizę wielopierwiastkową; wysoka temperatura plazmy pozwala na oznaczenie pierwiastków o wysokich potencjałach wzbudzenia (np. W, Cl, Br, I, S, U); duży zakres prostoliniowości wskazań obejmujący 4 – 5 rzędów wielkości stężenia; pozwala oznaczać zarówno składniki główne jak i śladowe w tej samej próbce; do wzbudzenia nie używa się elektrod (brak zanieczyszczeń); granica wykrywalności w zakresie 0,1 –10 μg/l; użycie polichromatora umożliwia oznaczenie ok. 60 pierwiastków w ciągu kilku minut; brak absorpcji wewnątrz źródła; brak zakłóceń związanych z obecnością tlenu;
; duży zakres prostoliniowości wskazań obejmujący 4 – 5 rzędów wielkości stężenia; pozwala oznaczać zarówno składniki główne jak i śladowe w tej samej próbce; do wzbudzenia nie używa się elektrod (brak zanieczyszczeń); granica wykrywalności w zakresie 0,1 –10 μg/l; użycie polichromatora umożliwia oznaczenie ok. 60 pierwiastków w ciągu kilku minut; brak absorpcji wewnątrz źródła; brak zakłóceń związanych z obecnością tlenu;.")
94
Wady techniki ICP-OES −wysokie koszty analizy; −konieczność stosowania argonu i innych odczynników o bardzo wysokiej czystości; −rozpiętość granic wykrywalności dla poszczególnych pierwiastków w zakresie −kilku rzędów wielkości (co utrudnia analizę wielopierwiastkową); −interferencje spektralne dla pierwiastków bogatych w linie emisyjne, takich jak: U, W, Co, Fe; −rutynowe operacje i naprawy muszą być wykonywane przez wysoce −wykwalifikowany personel; −występowanie interferencji między pierwiastkami o podobnych −długościach fali; −konieczność kontroli temperatury otoczenia i wilgotności; −konieczność bardzo dobrej monochromatyzacji; −trudności związane z oznaczeniem litowców.
; −interferencje spektralne dla pierwiastków bogatych w linie emisyjne, takich jak: U, W, Co, Fe; −rutynowe operacje i naprawy muszą być wykonywane przez wysoce −wykwalifikowany personel; −występowanie interferencji między pierwiastkami o podobnych −długościach fali; −konieczność kontroli temperatury otoczenia i wilgotności; −konieczność bardzo dobrej monochromatyzacji; −trudności związane z oznaczeniem litowców.")
95
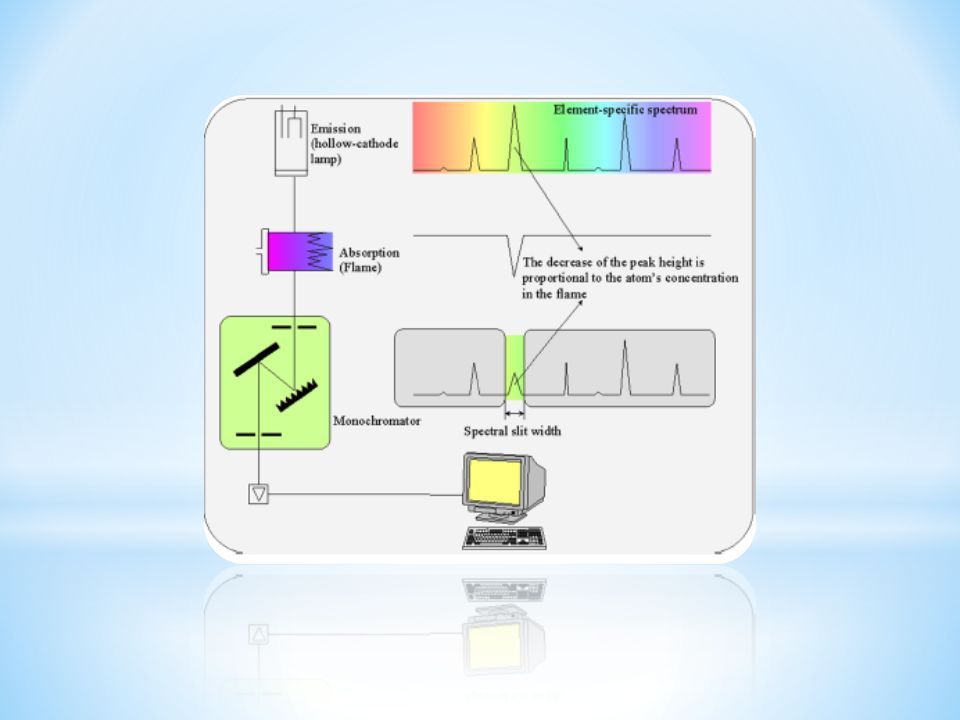
Spektroskopia absorpcji atomowej AAS Atomic Absorption Spectroscopy Wykorzystuje zjawisko absorpcji przez wolne atomy, charakterystycznych dla danego pierwiastka linii rezonansowych, emitowanych przez źródło promieniowania.

96
Schemat blokowy spektrometru AAS

97
Schemat spektrometru AAS

98
Schemat spektrometru dwukanałowgo AAS

101
Źródła promieniowania

103
Atomizery

105
W przypadku techniki zimnych par, rtęć jest uwalniana z badanej próbki a następnie (po ewentualnej redukcji do rtęci atomowej) jest zatrzymywana na złożu ze złotem w postaci amalgamatu. Następnie amalgamat jest podgrzewany do temperatury 600 °C a uwolniona atomowa rtęć jest kierowana w strumieniu powietrza do kuwety pomiarowej, gdzie następuje pomiar absorbancji dla charakterystycznej długości fali 253,7 nm. Źródłem promieniowania jest rtęciowa lampa z katodą wnękową.

106
Ten sposób pomiaru gwarantuje selektywność ponieważ: reakcja tworzenia amalgamatu jest selektywna dla rtęci, absorpcja promieniowania następuje przy długości fali charakterystycznej dla rtęci.

107
Przykłady oznaczeń

108
1. Procesy emisji: - specyficzna (cieplna i fluoroscencja atomowa), - niespecyficzna 2. Rozpraszanie promieniowania przez matrycę 3. Absorpcja cząsteczkowa Przyczyny zakłóceń w analizie AAS

109
1. Korekcja metodą ślepej próby lub z zastosowaniem materiałów odniesienia 2. Korekcja metodą dwóch linii. Spektrometr dwukanałowy. Na jednym kanale mierzy się sumę absorbcji atomowej, cząsteczkowej i rozpraszania, a na drugiej absorbcji cząsteczkowej i rozpraszania dla linii nie absorbowanej przez badane atomy (odległość spektralna linii jak najmniejsza - 0,5 - 5 nm). 3. Korekcja metodą Smith - Hieftje (High speed self rewersal method). Lampa z katodą wnękowa emituje normalne widmo (wąska linia) oraz w krótkich impulsach o dużym natężeniu rozdwojone widmo. Szeroki pik jest, przede wszystkim, absorbowany przez tło. 4. Korekcja z lampą deuterową. Na przemian lampy HCL lub ECL i lampy deuterowej (widmo ciągłe). Absorpcja na tej samej drodze. Korekcja tła
. 3. Korekcja metodą Smith - Hieftje (High speed self rewersal method). Lampa z katodą wnękowa emituje normalne widmo (wąska linia) oraz w krótkich impulsach o dużym natężeniu rozdwojone widmo. Szeroki pik jest, przede wszystkim, absorbowany przez tło. 4. Korekcja z lampą deuterową. Na przemian lampy HCL lub ECL i lampy deuterowej (widmo ciągłe). Absorpcja na tej samej drodze. Korekcja tła.")
110
5. Korekcja tła z wykorzystaniem efektu Zeemana (rozszczepienie poziomów wolnych atomów w zewnętrznym polu magnetycznym). Z jednej linii powstają trzy, leżące blisko siebie, ale odmiennie spolaryzowane. Składowa odpowiadająca pierwotnej długości służy do pomiaru absorpcji całkowitej, a pozostałe do absorpcji cząsteczkowej i rozpraszania. Wady: spadek czułości, wzrost szumu lampy HCL oraz przydatność tylko do atomizacji elektrotermicznej.
. Z jednej linii powstają trzy, leżące blisko siebie, ale odmiennie spolaryzowane. Składowa odpowiadająca pierwotnej długości służy do pomiaru absorpcji całkowitej, a pozostałe do absorpcji cząsteczkowej i rozpraszania. Wady: spadek czułości, wzrost szumu lampy HCL oraz przydatność tylko do atomizacji elektrotermicznej..")
111
Atomizacja w płomieniu (F-AAS) Zalety: - łatwy, bezpośredni pomiar; - stosunkowo niski koszt aparatury; - prosta obsługa; - krótki czas pomiaru (przy dobrze skalibrowanym aparacie); - dobra odtwarzalność. Wady −tylko część próbki ulega odparowaniu; −można analizować tylko roztwory rozcieńczone; −dość niska czułość pomiaru; −efekty matrycowe; −metoda oznaczania pojedynczych pierwiastków; −nie nadaje się do oznaczania składników głównych.

112
Atomizacja elektrotermiczna (ET-AAS) Zalety: wysoka czułość oznaczeń; można analizować próbki bez wstępnego przygotowania; można analizować próbki stałe; granice wykrywalności rzędu μg/l; możliwość analizy mikropróbek; duża selektywność Wady: −niska odtwarzalność; −metoda oznaczania pojedynczych pierwiastków; −efekty matrycowe
 Zalety: wysoka czułość oznaczeń; można analizować próbki bez wstępnego przygotowania; można analizować próbki stałe; granice wykrywalności rzędu μg/l; możliwość analizy mikropróbek; duża selektywność Wady: −niska odtwarzalność; −metoda oznaczania pojedynczych pierwiastków; −efekty matrycowe")
113
Granice oznaczalności: metoda płomieniowa - 1 – 1000 μg/dm 3 metoda bezpłomieniowa – 0,01 – 50 μg/dm3 (ppb)
")
114
Spektrofotometr AAS

Podobne prezentacje