Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
Zakład Genetyki Medycznej GENETYCZNIE UWARUNKOWANE CHOROBY METABOLICZNE: PRE- I POSTNATALNA DIAGNOSTYKA MICHAŁ PIETRUSIŃSKI ZAKŁAD GENETYKI MEDYCZNEJ UNIWERSYTET MEDYCZNY W ŁODZI ŁÓDŹ 2008

2
Zakład Genetyki Medycznej Zaburzenia przemiany aminokwasów fenyloketonuria klasyczna fenyloketonuria - postacie nietypowe alkaptonuria Zaburzenia przemiany puryn zespół Lescha-Nyhana Zaburzenia przemiany węglowodanowej galaktozemia Lizosomalne choroby spichrzeniowe mukopolisacharydozy typu I i II choroba Taya-Sachsa Mukowiscydoza

3
Zakład Genetyki Medycznej ZABURZENIA PRZEMIANY AMINOKWASÓW

4
Zakład Genetyki Medycznej HIPERFENYLOALANINEMIA Zaburzenia homeostazy fenyloalaniny - hydroksylaza fenyloalaninowa (97%) - reduktaza dwuhydropterynowa - synteza biopteryn dziedziczenie autosomalne recesywne
 - reduktaza dwuhydropterynowa - synteza biopteryn dziedziczenie autosomalne recesywne")
6
Zakład Genetyki Medycznej FENYLOKETONURIA KLASYCZNA PKU CZĘSTOŚĆ WYSTĘPOWANIA 1:2000 - 1-20000 1:8000 W POLSCE Brak HPA > wzrost st. Fenyloalaniny > kw. Fenylopirogronowy > kw. Ortohydroksyfenylooctowy Uszkodzenie OUN

7
Zakład Genetyki Medycznej OBJAWY KLINICZNE - uporczywe wymioty - zmiany skórne typu skazowego - mysi zapach moczu (kwas ortohydroksyfenylooctowy) - opóźnienie rozwoju psychoruchowego - opóźnienie rozwoju umysłowego - upośledzenie umysłowe znacznego stopnia - w 50% drgawki przed 1 r.ż. - małogłowie - jasna karnacja, blond włosy - rozcieńczenie barwnika

8
Zakład Genetyki Medycznej

9
- ilościowe oznaczenie fenyloalaniny i tyrozyny w surowicy krwi - ilościowe oznaczenie aktywności reduktazy dwuhydropterydynowej - ilościowe oznaczenie biopteryny i neopteryny w moczu (HPLC) U matek o pełnym profilu biochemicznym fenyloketonurii nieodwracalne uszkodzenia płodów
 U matek o pełnym profilu biochemicznym fenyloketonurii nieodwracalne uszkodzenia płodów")
10
Zakład Genetyki Medycznej NIETYOPWE POSTACIE FENYLOKETONURII Czterohydrobiopteryna > tryptofan > tyrozyna > fenyloalanina neurotransmitery: dopamina, serotonina - ciężkie napady drgawek - zahamowanie rozwoju psychoruchowego - zgon we wczesnym okresie życia

11
Zakład Genetyki Medycznej DIAGNOSTYKA MOLEKULARNA PAH - HYDROKSYLAZA FENYLOALANINOWA 12q23.2 90kb prawidłowy białkowy produkt - 452 aminokwasy 67% - missense

12
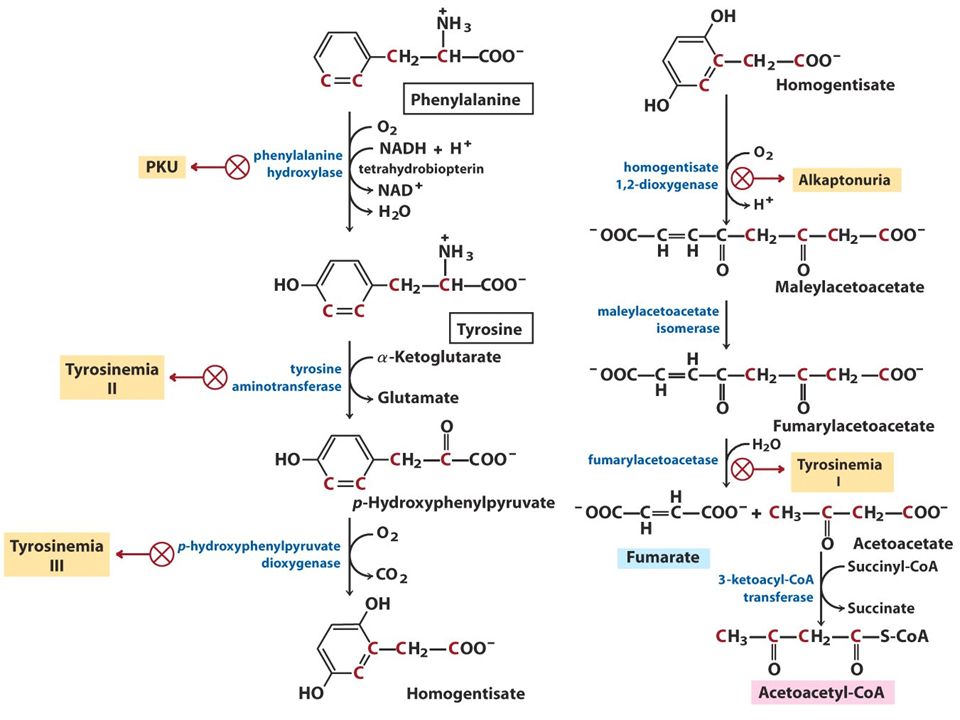
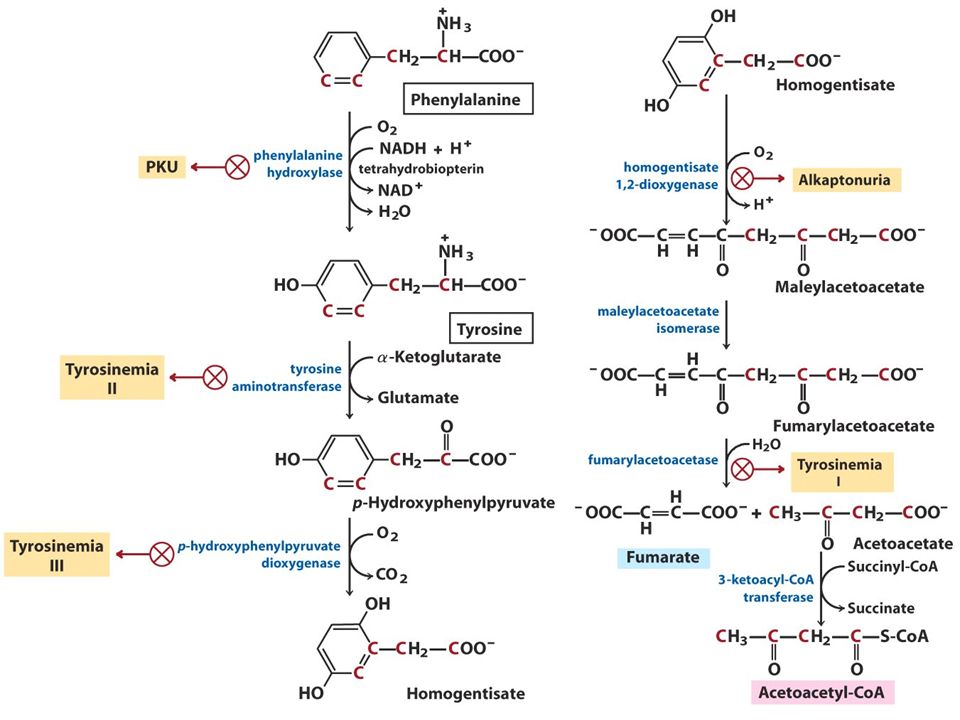
Zakład Genetyki Medycznej ALKAPTONURIA autosomalna recesywna Zaburzenie przemiany tyrozyny

14
Zakład Genetyki Medycznej u niemowląt i małych dzieci bezobjawowo ciemnienie pieluch po kontakcie z mydłem i powietrzem obecność kwasu homogentyzynowego w moczu II i III dekada – ochronoza-ciemnienie chrząstek, ścięgien, więzadeł oraz twardówek III i IV dekada - stany zapalne i zmiany zwyrodnieniowe stawów

15
Zakład Genetyki Medycznej DIAGNOSTYKA Oznaczenie zawartości HGA w moczu 1-8g dziennie 1mg Mutacje w genie HGD (1,2-dioksygenazy homogentyzynowej) 3q21-q23 14 egzonów 54,3kb transkrypt 1715bp znanych przynajmniej 67 mutacji, większość missense
 3q21-q23 14 egzonów 54,3kb transkrypt 1715bp znanych przynajmniej 67 mutacji, większość missense")
16
Zakład Genetyki Medycznej ZABURZENIA PRZEMIANY PURYN

17
Zakład Genetyki Medycznej ZESPÓŁ LESHA-NYHANA Recesywna sprzężona z chromosomem X Brak aktywności HGPRT (transferazy fosforybozylo-hipoksantyno-guaninowej)
")
18
Zakład Genetyki Medycznej Zasady purynowe 5-fosfozylofosforan (PRPP) nukleotydy Rybozo-5-fosforan HGPRT
 nukleotydy Rybozo-5-fosforan HGPRT")
19
Zakład Genetyki Medycznej Objawy kliniczne Od około 6 m-ca zycia - zaburzenia chodu - spastyczność - nadmiernie żywe odruchy - opóźniony rozwój - opistotonus - zmiany stawowe - zaburzenia psychiczne: autoagresja, samookaleczenia - zaburzenia czynności nerek - hiperurikemia > 535μmol/l częściowy blok - brak objawów neurologicznych

20
Zakład Genetyki Medycznej diagnostyka - stosunek kwasu moczowego do kreatyniny > 2 - >20mg kwasu moczowego/doba (charakterystyczne ale nie wydaje się na tej podstawie diagnozy) - hiperurikemia(j.w) - aktywność HGPRT w wielu tkankach (krew, fibroblasty) poniżej 1,5%
 - hiperurikemia(j.w) - aktywność HGPRT w wielu tkankach (krew, fibroblasty) poniżej 1,5%")
21
Zakład Genetyki Medycznej HGPRT Xq26-q27.2 43kb

22
Zakład Genetyki Medycznej ZABURZENIA PRZEMIANY WĘGLOWODANOWEJ

23
Zakład Genetyki Medycznej Zaburzenie metabolizmu galaktozy 1:1700 do 1:50000 żywo urodzonych autosomalna, recesywna GALAKTOZEMIA

24
galaktoza galaktozo-1-fosforan urydylotransferaza galaktozo-1-fosforanowa Urydynodwufosfoglukoza (UDPG) Urydynodwufosfogalaktoza (UDPGal) Glukozo-1-fosforan galaktokinaza epimeraza UDP glukozowa glikolipidy ATP glikoproteidywielocukry
 Urydynodwufosfogalaktoza (UDPGal) Glukozo-1-fosforan galaktokinaza epimeraza UDP glukozowa glikolipidy ATP glikoproteidywielocukry")
25
Zakład Genetyki Medycznej BRAK AKTYWNOŚCI URYDYLOTRANSFERAZY GALAKTOZO-1-FOSFORANOWEJ - objawy kliniczne U noworodków: - biegunka - wymioty - niechęć ssania - kwasica - utrata masy ciała - pocenie się - senność - hipoglikemia - wrażliwość na zakażenia - szczególnie E.coli obraz sugeruje infekcje

26
Zakład Genetyki Medycznej Wątroba - niewydolność wątroby - hepatomegalia lub hepatosplenomegalia - żółtaczka - hipoalbuminemia - hipoprotrombinemia Nerki - leukocyturia - erytrocyturia - proteinuria - glikozuria - hiperaminocyturia Uszkodzenie OUN - opóźnienie rozwoju psychomotorycznego, zmiany w zapisie EEG, uszkodzenie soczewki oka - zaćma

27
Zakład Genetyki Medycznej Gen urydylotransferazy galaktozo-1-fosforanowej 9p13 4kb 11 egzonów 8 najczęściej występujących mutacji - 71%

28
Zakład Genetyki Medycznej LIZOSOMALNE CHOROBY SPICHRZENIOWE

29
Zakład Genetyki Medycznej Niedobór aktywności specyficznych enzymów lizosomalnych > gromadzenie w lizosomach wielkocząsteczkowych związków - sfingolipidy, mukopolisacharydy, glikoproteidy itp... Znanych około 30 chorób tego typu klasyfikacja wg rodzaju substratu przeważającego w złogach

30
Zakład Genetyki Medycznej MUKOPOLISACHARYDOZY Deficyt hydrolaz lub sulfataz zajęcie układu nerwowego, narządów miąższowych, układu kostnego, narządu wzroku, tk. łącznej. 13 typów 1:20000

31
Zakład Genetyki Medycznej MUKOPOLISACHARYDOZA TYP I (MPS I) CHOROBA HURLER - gargoidalne rysy twarzy - powiekszenie obwodu głowy - przykurcze w stawach - skrócenie i pogrubienie kości - powiekszenie śledziony i wątroby - zmętnienie rogówki - niedosłuch - przepuklina pępkowa - postępująca degradacja umysłowa - zgon w II dekadzie życia Autosomalna recesywna
 CHOROBA HURLER - gargoidalne rysy twarzy - powiekszenie obwodu głowy - przykurcze w stawach - skrócenie i pogrubienie kości - powiekszenie śledziony i wątroby - zmętnienie rogówki - niedosłuch - przepuklina pępkowa - postępująca degradacja umysłowa - zgon w II dekadzie życia Autosomalna recesywna")
32
Zakład Genetyki Medycznej DIAGNOSTYKA wstępnie Ilościowe i jakościowe oznaczenie w moczu glikozaminoglikanów (GAG): heparan, siarczan dermatanu Wykazanie niedostatecznej aktywności α-L-iduronidazy. Aktywność mierzona w limfocytach krwi obwodowej, fibroblastach (hodowle), osoczu, amniocytach (hodowle) analiza mutacji w genie IDUA
, osoczu, amniocytach (hodowle) analiza mutacji w genie IDUA.")
33
Zakład Genetyki Medycznej Gen α-L-iduronidazy - IDUA 4p16.3 14 egzonów 19 kb znanych ponad 60 mutacji

34
Zakład Genetyki Medycznej HURLER-SCHEIE SYNDROM - MPS I POŚREDNIA SCHEIE SYNDROM - MPS I ŁAGODNA Prawidłowy rozwój do 24 miesięcy symptomy zazwyczaj między 3 a 8 rokiem życia często dożywalność do wieku dojrzałego intelekt zróżnicowany często całkowicie prawidłowy Diagnozowana często po 15 roku życia prawidłowy intelekt prawidłowa postawa Zwyrodnienie stawów Zaburzenia zastawek serca Przerost narządów wewnętrznych

35
Zakład Genetyki Medycznej MUKOPOLISACHARYDOZA TYP II (MPS II) CHOROBA HUNTERA Recesywna sprzężona z chromosomem X (Xq28) Objawy jak zespole Hurler występują nieco później brak zmętnienia rogówki Typ A szybko postępujące upośledzenie umysłowe pogrubione rysy twarzy, zgon w II dekadzie życia Typ B rozwój umysłowy w granicach normy osiagnięcie wieku dojrzałego
 CHOROBA HUNTERA Recesywna sprzężona z chromosomem X (Xq28) Objawy jak zespole Hurler występują nieco później brak zmętnienia rogówki Typ A szybko postępujące upośledzenie umysłowe pogrubione rysy twarzy, zgon w II dekadzie życia Typ B rozwój umysłowy w granicach normy osiagnięcie wieku dojrzałego")
36
Zakład Genetyki Medycznej MUKOWISCYDOZA

37
Zakład Genetyki Medycznej MUKOWISCYDOZA CYSTIC FIBROSIS Torbielowate zwłóknienie trzustki

38
Zakład Genetyki Medycznej PATOGENEZA Wzmożona lepkość wydzieliny gruczołów wydzielania zewnętrznego > zaczopowanie przewodów wyprowadzających - trzustka - wątroba - jelita - nasieniowody - płuca - gruczoły potowe

39
Zakład Genetyki Medycznej CZĘSTOŚĆ WYSTĘPOWANIOA 1:1800 - 1:2500 1:2300 W POLSCE MONOGENOWA AUTOSOMALNA RECESYWNA

40
Zakład Genetyki Medycznej OBJAWY KLINICZNE Postać płucna - przyspieszony oddech, kaszel, infekcje Postać brzuszna - zaburzenia trawienia i wchłaniania, cuchnące i tłuszczowe stolce, wypadanie odbytnicy, zapalenie trzustki i pęcherzyka żółciowego, cukrzyca - łagodna postać Postać mieszana

41
Zakład Genetyki Medycznej BIAŁKO CFTR ABC Family (ATP-binding casette) Kanał chlorkowy zależny od cAMP 1488aa 170kDa CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR
 Kanał chlorkowy zależny od cAMP 1488aa 170kDa CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR")
42
Zakład Genetyki Medycznej GEN CFTR 7q31.3 27 egzonów 250kb

43
Zakład Genetyki Medycznej DIAGNOSTYKA MUTACJI delF508 - analiza wielkości produktu PCR Del2,3(21kb) - analiza wielkości produktu PCR 3849+10kb - analiza fragmentów restrykcyjnych
 - analiza wielkości produktu PCR kb - analiza fragmentów restrykcyjnych")
44
PCR DNA startery dNTPpolimeraza bufor woda TERMOCYKLER

45
Zakład Genetyki Medycznej DENATURACJA 95°C

46
Zakład Genetyki Medycznej DENATURACJA 95°C

47
Zakład Genetyki Medycznej WIĄZANIE STARTERÓW °C

48
Zakład Genetyki Medycznej ELONGACJA 72°C

49
delF508

50
Zakład Genetyki Medycznej 1 2 3

51
2 3 1

52
Dele2,3 delecja 21kb

53
3849+10kb C>T PCR Trawienie Hph1

Podobne prezentacje





 nerek>")








 i WTÓRNE (NABYTE) NIEDOBORY ODPORNOŚCI>")




