Pobierz prezentację
Pobieranie prezentacji. Proszę czekać

1
CHEMIA TEORETYCZNA N. Smirnova – Metody termodynamiki statystycznej w chemii fizycznej J. Stecki – Termodynamika statystyczna K. Gumiński, P. Petelenz – Elementy chemii teoretycznej

2
Teorie w naukach przyrodniczych można podzielić na: - fenomenologiczne (opisowe) - modelowe
Te dwa nurty (fenomenologiczny i modelowy) łączy w sobie: - w obszarze zjawisk atomowych – mechanika kwantowa - dla zjawisk makroskopowych – mechanika statystyczna Podstawy tej ostatniej położyli pod koniec XIX w Ludwik Boltzmann i Jozajasz Willard Gibbs.
 łączy w sobie: - w obszarze zjawisk atomowych – mechanika kwantowa. - dla zjawisk makroskopowych – mechanika statystyczna. Podstawy tej ostatniej położyli pod koniec XIX w Ludwik Boltzmann i Jozajasz Willard Gibbs.")
3
Pojęcia wstępne: W termodynamice chemicznej rozważa się zawsze bądź reakcje chemiczne, bądź procesy fizykochemiczne (np. parowanie, rozpuszczanie, solwatację, itp.) przebiegające w naczyniu o pewnej objętości V, pod określonym ciśnieniem p, i w temperaturze T. Przestrzeń, w której zachodzi rozważany proces wraz ze znajdującymi się tam substancjami to układ. Zmienne określające stan układu (V, p, T, ni – skład) – parametry stanu. Parametry stanu, których wielkość zależy od masy substancji zawartej w układzie (np. V – objętość układu) są parametrami ekstensywnymi. Parametry stanu, których wielkość nie zależy od masy substancji zawartej w układzie (np. T) są parametrami intensywnymi. Wszystko to, co znajduje się poza układem, określa się jako otoczenie układu.
 przebiegające w naczyniu o pewnej objętości V, pod określonym ciśnieniem p, i w temperaturze T. Przestrzeń, w której zachodzi rozważany proces wraz ze znajdującymi się tam substancjami to układ. Zmienne określające stan układu (V, p, T, ni – skład) – parametry stanu. Parametry stanu, których wielkość zależy od masy substancji zawartej w układzie (np. V – objętość układu) są parametrami ekstensywnymi. Parametry stanu, których wielkość nie zależy od masy substancji zawartej w układzie (np. T) są parametrami intensywnymi. Wszystko to, co znajduje się poza układem, określa się jako otoczenie układu.")
4
Typy układów: Jeżeli ścianki odgradzające układ od otoczenia nie przepuszczają ani masy ani energii, to taki układ nazywamy izolowanym. Parametry ustalone to E,V,N. Jeżeli pomiędzy układem a otoczeniem nie ma wymiany masy, to układ określamy jako zamknięty. Parametry ustalone T,V,N. Natomiast, gdy przez ścianki oddzielające przenika masa, układ jest otwarty. Parametry ustalone µ,T,V.

5
Termodynamika fenomenologiczna – teoria materii, której podstawowe pojęcia pochodzą bezpośrednio z doświadczeń. Jej podstawowe prawa zostały sformułowane w wyniku uogólnienia danych uzyskiwanych w doświadczeniach makroskopowych. Prawa te podają ogólne zależności pomiędzy wielkościami makroskopowymi, które są spełnione dla każdego układu – niezależnie od rodzaju tworzących go cząstek. Prawa te nie dają jednak możliwości interpretacji teoretycznej właściwości układu, które zależą od rodzaju tworzących go składników. Np. termodynamiczny warunek równowagi chemicznej w układzie, w który przebiega reakcja chemiczna dana równaniem reakcji: ma postać: ale nie możemy obliczyć stałej równowagi, bo nie znamy potencjałów standardowych.

6
MIKROSTAN I MAKROSTAN UKŁADU Rozważmy układ złożony np
MIKROSTAN I MAKROSTAN UKŁADU Rozważmy układ złożony np. z N cząsteczek gazu, zajmujący w temperaturze T objętość V. Cząsteczki gazu znajdują się w nieustannym ruchu i rozważany układ znajduje się w coraz to nowych stanach dynamicznych. Gdyby udało nam się ustalić w danej chwili położenia i pędy wszystkich cząsteczek układu, to znalibyśmy mikrostan układu. Jest on oczywiście niedostępny bezpośrednim pomiarom. Doświadczalnie możemy badać jedynie makrostan układu, który jest określany za pomocą makroskopowych parametrów stanu (p, T, V). Wartości parametrów stanu nie pozwalają nic powiedzieć o położeniu poszczególnych cząsteczek w układzie. Np. ciśnienie wywierane na ścianki naczynia nie zależy od tego, która cząsteczka przekazuje ściance swój pęd, ale jedynie ile cząsteczek i o jakiej prędkości dokonało tego. Tak więc danemu makrostanowi odpowiada bardzo dużo różnych mikrostanów. Im więcej różnych mikrostanów odpowiada danemu makrostanowi, tym większe jest prawdopodobieństwo wystąpienia danego makrostanu.
. Wartości parametrów stanu nie pozwalają nic powiedzieć o położeniu poszczególnych cząsteczek w układzie. Np. ciśnienie wywierane na ścianki naczynia nie zależy od tego, która cząsteczka przekazuje ściance swój pęd, ale jedynie ile cząsteczek i o jakiej prędkości dokonało tego. Tak więc danemu makrostanowi odpowiada bardzo dużo różnych mikrostanów. Im więcej różnych mikrostanów odpowiada danemu makrostanowi, tym większe jest prawdopodobieństwo wystąpienia danego makrostanu.")
7
Stan mechaniczny zbioru cząstek o f stopniach swobody Do pełnego określenia stanu jakiegoś układu konieczna jest w mechanice klasycznej znajomość: - wszystkich współrzędnych określających położenie cząstek - wszystkich pędów odpowiadających tym współrzędnym. Liczba stopni swobody f – najmniejsza liczba współrzędnych potrzebna do określenia położenia cząstki w sposób wystarczający (jednoznaczny). Liczba stopni swobody układu N cząstek: F=Nf. Liczba wielkości potrzebnych do pełnego określenia stanu mechanicznego cząstki: 2f. Liczba wielkości (zmiennych niezależnych) potrzebnych do pełnego określenia stanu mechanicznego: 2F. Zmienne niezależne, którymi opisujemy stan układu nazywamy zmiennymi kanonicznymi. q – współrzędne uogólnione, p – pędy uogólnione
. Liczba stopni swobody układu N cząstek: F=Nf. Liczba wielkości potrzebnych do pełnego określenia stanu mechanicznego cząstki: 2f. Liczba wielkości (zmiennych niezależnych) potrzebnych do pełnego określenia stanu mechanicznego: 2F. Zmienne niezależne, którymi opisujemy stan układu nazywamy zmiennymi kanonicznymi. q – współrzędne uogólnione, p – pędy uogólnione.")
8
Przestrzeń fazowa q11, q21, …, qf1, q12, …, qf2, …, q1N, …, qfN, p11, p21, …, pf1, p12, …, pf2, …, p1N, …, pfN współrzędne pierwszej cząstki W przestrzeni tej rozpatrujemy układ współrzędnych o 2Nf osiach. Na każdej osi nanosimy jedną z 2Nf wielkości określających stan układu. Wyznaczy to w tej przestrzeni pewien punkt. Punkt ten przedstawia jednoznacznie stan układu mechanicznego złożonego z N cząstek – nazywamy go punktem fazowym układu. Przestrzeń 2Nf wymiarową nazywamy przestrzenią fazową . Np. dla układu N atomów w R3: przestrzeń jest 6N wymiarowa: 1 atom: x1, y1, z1, px1, py1, pz1 N atomów: x1, y1, z1, x2, y2, z2, …, xN, yN, zN, px1, py1, pz1, px2, py2, pz2, …, pxN, pyN, pzN

9
Przestrzeń fazowa dla 3 cząstek w przestrzeni jednowymiarowej (R1)
x1 N=3 ; R1 (x1,px1) (x2,px2) (x3,px3) px3 px1 x3 px2 x2
 (x2,px2) (x3,px3) px3. px1. x3. px2. x2.")
10
Przestrzeń fazowa Podobnie można pomyśleć o przestrzeni jednej cząstki o 2f wymiarach (f współrzędnych i f pędów). Stan cząstki jest określony przez jeden punkt w tej przestrzeni. Otrzymany punkt nazywamy punktem fazowym cząstki. Przedstawia jednoznacznie stan mechaniczny tej cząstki. Przestrzeń nazywa się przestrzenią fazową . Gdybyśmy w przestrzeni m przedstawili stany wszystkich N cząstek gazu, to otrzymalibyśmy w niej N punktów. W przestrzeni stan układu byłby przedstawiony jednym punktem. px x o o o o ooo o o o Trajektoria fazowa Położenie punktu fazowego zmienia się w czasie. Punkt ten porusza się – zakreśla krzywą zwaną trajektorią fazową.

11
STAN MAKRO I STAN MIKRO ________________________________________________________ Każda zmiana współrzędnych czy pędów poszczególnych cząstek (a także wymiana współrzędnych i pędów między dwiema identycznymi cząstkami) spowoduje zmianę położenia punktu fazowego w przestrzeni . Natomiast taka wymiana pozostawi to samo rozmieszczenie liczbowe punktów fazowych poszczególnych cząstek w różnych komórkach przestrzeni m. Makroskopowe własności gazu nie zależą od tego, które cząstki mają takie a takie współrzędne i takie a takie pędy. Jedynie od tego ile cząstek ma takie a takie współrzędne, takie a takie pędy. Zatem makroskopowe własności gazu zależą od tego, jakie jest rozmieszczenie punktów fazowych w różnych komórkach przestrzeni fazowej . Stan makro układu – określone rozmieszczenie liczbowe cząstek pomiędzy różne wartości współrzędnych i pędów. Stan mikro układu – dokładne podanie współrzędnych i pędów poszczególnych cząstek. Stan makro może być realizowany przez cały szereg stanów mikro.

12
Mechanika statystyczna klasyczna i mechanika statystyczna kwantowa
Mechanika statystyczna kwantowa różni się od klasycznej w dwu założeniach, mimo że obie wynikają z fundamentalnych postulatów mechaniki kwantowej i mają swój początek w zasadzie nieoznaczoności Heisenberga: Pierwsza różnica: Stan układu w mechanice klasycznej jest dany poprzez dokładne określenie wszystkich współrzędnych i wszystkich pędów cząstek wchodzących w skład układu, natomiast w mechanice kwantowej stan układu dany jest przez określenie funkcji falowej zależącej tylko od współrzędnych cząstek. Stan N cząstek o f stopniach swobody: Klasycznie - Kwantowo - Druga różnica: W mechanice klasycznej identyczne cząstki są rozróżnialne, a w mechanice kwantowej nierozróżnialne. W mechanice kwantowej stanem mikro układu jest stan kwantowo-mechaniczny całego układu wyrażony funkcją falową zbudowaną z funkcji jednocząstkowych. Takim funkcjom własnym odpowiadają odpowiednie wartości własne energii cząstki (kwantowane). Odpowiednikiem stanu makro jest określone liczbowe przyporządkowanie cząstek poszczególnym poziomom energetycznym. q11, q21, …, qf1, q12, …, qf2, …, q1N, …, qfN, p11, p21, …, pf1, p12, …, pf2, …, p1N, …, pfN
. Odpowiednikiem stanu makro jest określone liczbowe przyporządkowanie cząstek poszczególnym poziomom energetycznym. q11, q21, …, qf1, q12, …, qf2, …, q1N, …, qfN, p11, p21, …, pf1, p12, …, pf2, …, p1N, …, pfN.")
13
Przykłady trajektorii fazowych: Przykład pierwszy: Trajektoria fazowa cząstki poruszającej się ruchem jednostajnym prostoliniowym wzdłuż osi x, px=const. px x Przykład drugi: Jednowymiarowy oscylator harmoniczny. -A A kołowa częstość drgań siła Hooka k – stała siłowa x

14
p x e1 e2 e1< e2

15
Obliczanie średnich wartości wielkości mechanicznych
M(p(t),q(t)) – pewna funkcja uogólnionych pędów i współrzędnych t – czas trwania doświadczenia Mt – średnia wartość M – średnia czasowa, czyli średnia po trajektorii fazowej zakreślonej przez punkt fazowy układu w danym czasie dt(p,q) – czas, w ciągu którego punkt fazowy układu znajduje się w elemencie objętości dpdq wokół punktu o współrzędnych p i q. Metoda ta napotyka szereg trudności (między innymi rachunkowych, stosowana tylko dla małej ilości cząstek). Ponadto podstawowe parametry termodynamiczne jak temperatura, entropia czy potencjał chemiczny nie są średnimi wartościami wielkości mechanicznych, więc nie można ich obliczać z powyższego wzoru.
,q(t)) – pewna funkcja uogólnionych pędów i współrzędnych. t – czas trwania doświadczenia. Mt – średnia wartość M – średnia czasowa, czyli średnia po trajektorii fazowej zakreślonej przez punkt fazowy układu w danym czasie. dt(p,q) – czas, w ciągu którego punkt fazowy układu znajduje się w elemencie objętości dpdq wokół punktu o współrzędnych p i q. Metoda ta napotyka szereg trudności (między innymi rachunkowych, stosowana tylko dla małej ilości cząstek). Ponadto podstawowe parametry. termodynamiczne jak temperatura, entropia czy potencjał chemiczny nie są. średnimi wartościami wielkości mechanicznych, więc nie można ich obliczać. z powyższego wzoru.")
16
Metoda zespołów Gibbsa
Zadaniem molekularnej teorii procesów makroskopowych jest wyjaśnienie zachowania się układów na podstawie praw mikroświata. Przedmiotem rozważań powinna być zmiana stanu układu w czasie (fazowa trajektoria układu) ale z wcześniej wymienionych powodów być nie może. Metody statystyczne stosuje się, gdy mamy niewystarczającą znajomość danego układu by móc z całkowitą pewnością przewidzieć jego przyszłe zachowanie się. W tym celu wykorzystujemy metodę zespołów statystycznych Gibbsa. Wprowadzamy probabilistyczny opis mikrostanów układu, tzn. traktujemy zmienne dynamiczne q i p jako wielkości przypadkowe i przypisujemy im (postulujemy) pewne prawdopodobieństwa wystąpienia. Zespół statystyczny – jest to zbiór bardzo dużej liczby identycznych układów, posiadających te same wartości parametrów makroskopowych (np. E, N, V, T, p…), znajdujących się w takich samych warunkach zewnętrznych i różniących się jedynie stanami mikroskopowymi. Poszczególne układy zbudowane są z cząstek tego samego rodzaju. Ich oddziaływania z otoczeniem są takie same. Parametry zewnętrzne oraz inne wielkości makroskopowe są jednakowe dla wszystkich układów zespołu.
 ale z wcześniej wymienionych powodów być nie może. Metody statystyczne stosuje się, gdy mamy niewystarczającą znajomość danego układu by móc z całkowitą pewnością przewidzieć jego przyszłe zachowanie się. W tym celu wykorzystujemy metodę zespołów statystycznych Gibbsa. Wprowadzamy probabilistyczny opis mikrostanów układu, tzn. traktujemy zmienne dynamiczne q i p jako wielkości przypadkowe i przypisujemy im (postulujemy) pewne prawdopodobieństwa wystąpienia. Zespół statystyczny – jest to zbiór bardzo dużej liczby identycznych układów, posiadających te same wartości parametrów makroskopowych (np. E, N, V, T, p…), znajdujących się w takich samych warunkach zewnętrznych i różniących się jedynie stanami mikroskopowymi. Poszczególne układy zbudowane są z cząstek tego samego rodzaju. Ich oddziaływania z otoczeniem są takie same. Parametry zewnętrzne oraz inne wielkości makroskopowe są jednakowe dla wszystkich. układów zespołu.")
17
Zespół statystyczny Gibbsa: zbiór identycznych układów znajdujących się w takich samych warunkach zewnętrznych i charakteryzowanych przez takie same wartości wybranych parametrów fizycznych (ten sam stan makroskopowy), ale różniących się stanami mikroskopowymi. Np..: N,T,V N,T,V N,T,V N,T,V N,T,V N,T,V N,T,V N,T,V T OTOCZENIE Układy zespołu reprezentują mikrostany, w których może się znaleźć nasz układ. takie same N,T, V różne (p,q) = różne mikrostany
 = różne mikrostany.")
18
E,V,N =const. N,V,T =const. V,T,µ =const.

19
prawdopodobieństwo gęstość prawdopodobieństwa mikrostanów

20
gęstość prawdopodobieństwa
prawdopodobieństwo

22
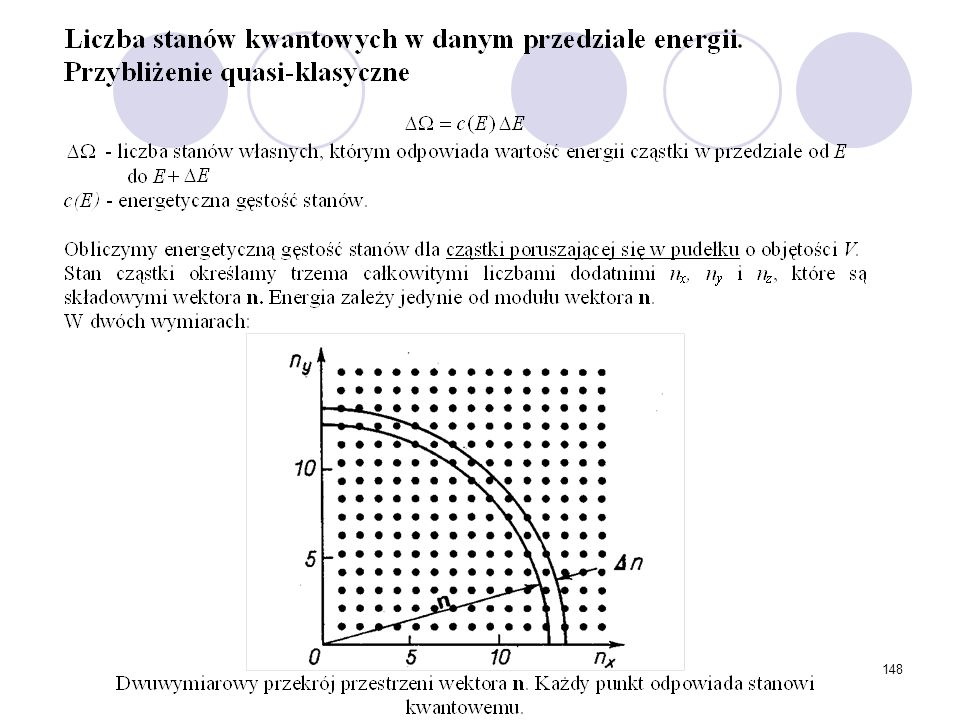
………… ZESPOŁY STATYSTYCZNE Zespół mikrokanoniczny (E,V,N)
Jest to zespół układów izolowanych. Dla każdego z układów zadajemy następujące parametry: energię E, liczbę cząstek N, objętość V. Ze względów rachunkowych, aby uzyskać funkcję rozkładu zakładamy, że energia zmienia się w pewnym wąskim (nieskończenie małym) przedziale . Przybliżenie takie ma swoje uzasadnienie w rzeczywistości doświadczalnej, gdyż nie jest możliwa całkowita izolacja energetyczna badanego układu. E,V,N …………
 przedziale . Przybliżenie takie ma swoje uzasadnienie w rzeczywistości doświadczalnej, gdyż nie jest możliwa całkowita izolacja energetyczna badanego układu. E,V,N. …………")
23
Warunek unormowania funkcji :
Prawo jednakowego prawdopodobieństwa stanów mikro o tej samej energii pozwala nam zdefiniować rozkład mikrokanoniczny: tzn. wszystkie stany leżące wewnątrz zadanej powłoki energetycznej są jednakowo prawdopodobne, a prawdopodobieństwo występowania stanów poza powłoką jest równe zeru. Warunek unormowania funkcji : . - objętość przestrzeni fazowej (powłoki energetycznej) odpowiadającej przedziałowi energii
 odpowiadającej przedziałowi energii.")
24
Dla układów makroskopowych objętość fazowa (E) jest szybko rosnącą funkcją energii. Dla N cząsteczek gazu doskonałego w objętości V o energii całkowitej ≤E: (E) E E+E
 E. E+E. ")
25
Prawdopodobieństwo stanu makro w(E,V,N,X) – podejście klasyczne
Jeżeli określająca stan makro zmienna losowa X jest zmienną normalną w sensie statystyczno–termodynamicznym, tzn. spełnione są warunki: wartość średnia jest równocześnie wartością najbardziej prawdopodobną (funkcja rozkładu jest symetryczna); fluktuacje zmiennej X są małe w porównaniu z wartością średnią (funkcja rozkładu jest bardzo wąska); to maksimum prawdopodobieństwa w(X) jest bardzo wąskie i stanowi, w którym X= X* odpowiada prawie cała objętości powłoki energetycznej. W(X) X* X Obszary powłoki energetycznej odpowiadające różnym makrostanom.
; fluktuacje zmiennej X są małe w porównaniu z wartością średnią (funkcja rozkładu jest bardzo wąska); to maksimum prawdopodobieństwa w(X) jest bardzo wąskie i stanowi, w którym X= X* odpowiada prawie cała objętości powłoki energetycznej. W(X) X* X. Obszary powłoki energetycznej odpowiadające różnym makrostanom.")
26
Możemy więc stwierdzić, że:
każdy stan makroskopowy układu można scharakteryzować za pomocą objętości fazowej odpowiadającej temu stanowi; objętość fazowa (lub ) jest funkcją stanu, tzn. zależy od parametrów charakterystycznych dla stanu układu (E,V,N,X), nie zależy od przeszłości układu; stan równowagowy jest stanem, któremu odpowiada prawie cała objętość powłoki energetycznej. Dla układu izolowanego Boltzmann wprowadził funkcję S: k – współczynnik proporcjonalności. Zauważmy, że: wielkość S jest funkcją stanu, ponieważ jest funkcją stanu; wielkość S rośnie wraz ze wzrostem prawdopodobieństwa, ponieważ jest proporcjonalna do prawdopodobieństwa rozpatrywanego makrostanu układu; wielkość S osiąga maksimum dla układu równowagowego, gdyż stanowi równowagi odpowiada największa wartość . Te własności funkcji S pokrywają się z własnościami funkcji stanu – entropii. Dlatego też powyższy wzór to statystyczna definicja entropii.
 jest funkcją stanu, tzn. zależy od parametrów charakterystycznych dla stanu układu (E,V,N,X), nie zależy od przeszłości układu; stan równowagowy jest stanem, któremu odpowiada prawie cała objętość powłoki energetycznej. Dla układu izolowanego Boltzmann wprowadził funkcję S: k – współczynnik proporcjonalności. Zauważmy, że: wielkość S jest funkcją stanu, ponieważ jest funkcją stanu; wielkość S rośnie wraz ze wzrostem prawdopodobieństwa, ponieważ jest proporcjonalna do prawdopodobieństwa rozpatrywanego makrostanu układu; wielkość S osiąga maksimum dla układu równowagowego, gdyż stanowi równowagi odpowiada największa wartość . Te własności funkcji S pokrywają się z własnościami funkcji stanu – entropii. Dlatego też powyższy wzór to statystyczna definicja entropii.")
27
Aby zapewnić zgodność pomiędzy statystyczną definicją entropii i definicją entropii w termodynamice fenomenologicznej należy przyjąć: k – stała Boltzmanna. Absolutne wartości entropii określa się w termodynamice za pomocą eksperymentalnych pomiarów kalorymetrycznych i trzeciego prawa termodynamiki. Aby zależności statystyczne dawały absolutną wartość entropii należy unormować objętość fazową za pomocą mnożnika mającego wymiar [działanie]-F.

28
II. Podejście kwantowo–mechaniczne
Absolutną wartość entropii można otrzymać jedynie za pomocą pojęć kwantowo-mechanicznych (wyrażając entropię przez liczbę stanów kwantowych realizujących dany stan makroskopowy). Jeżeli rozpatrujemy zbiór stanów dyskretnych wówczas entropię definiujemy za pomocą postulatu Boltzmanna: W – liczba mikrostanów realizujących dany stan makroskopowy (jest to prawdopodobieństwo termodynamiczne tego makrostanu). Dla układów zespołu mikrokanonicznego (izolowanych) w stanie równowagi entropia osiąga maksimum. Funkcję, która w stanie równowagi osiąga maksimum lub minimum nazywamy potencjałem termodynamicznym.
. Jeżeli rozpatrujemy zbiór stanów dyskretnych wówczas entropię definiujemy za pomocą postulatu Boltzmanna: W – liczba mikrostanów realizujących dany stan makroskopowy (jest to prawdopodobieństwo termodynamiczne tego makrostanu). Dla układów zespołu mikrokanonicznego (izolowanych) w stanie równowagi entropia osiąga maksimum. Funkcję, która w stanie równowagi osiąga maksimum lub minimum nazywamy potencjałem termodynamicznym.")
29
Związek objętości fazowej (sumy stanów) z funkcjami termodynamicznymi układu
Dla układów klasycznych objętość fazowa (E,V,N) (lub (E,V,N) ) jest podstawową wielkością liczoną w zespole mikrokanonicznym. Dla układów kwantowych odpowiednikiem (E) jest liczba mikrostanów W dla ustalonych E, V i N. Entropia układu znajdującego się w określonym stanie makroskopowym jest wielkością proporcjonalną do logarytmu wagi statystycznej (prawdopodobieństwa) tego stanu. To jest statystyczna definicja entropii Boltzmanna. Dla układów zespołu mikrokanonicznego (izolowanych) w stanie równowagi entropia osiąga maksimum. Funkcję, która w stanie równowagi osiąga maksimum lub minimum nazywamy potencjałem termodynamicznym. 29
 (lub (E,V,N) ) jest podstawową wielkością liczoną w zespole mikrokanonicznym. Dla układów kwantowych odpowiednikiem (E) jest liczba mikrostanów W dla ustalonych E, V i N. Entropia układu znajdującego się w określonym stanie makroskopowym jest wielkością proporcjonalną do logarytmu wagi statystycznej (prawdopodobieństwa) tego stanu. To jest statystyczna definicja entropii Boltzmanna. Dla układów zespołu mikrokanonicznego (izolowanych) w stanie równowagi entropia osiąga maksimum. Funkcję, która w stanie równowagi osiąga maksimum lub minimum nazywamy potencjałem termodynamicznym. 29.")
30
S p H - - U G + + F V T Schemat obliczeń w zespole mikrokanonicznym:
Obliczamy unormowaną objętość fazową: =d q1dp1dq2dp2…dqFdpF; 2. Obliczamy entropię: Przybliżenie Stirlinga dla bardzo dużych N > 106: Z zależności termodynamicznych znajdujemy pozostałe funkcje termodynamiczne i parametry układu. 4. Ustalamy związki pomiędzy parametrami opisującymi układ. S p H - - U G + + F V T

31
Przykład I (z poprzedniego wykładu !!!!)
N cząsteczek gazu doskonałego znajduje się w izolowanym naczyniu o objętości V. Wyprowadzić równanie stanu gazu doskonałego. Szukamy: f(p,V,T,N) = E = const.
 = 0 E = const.")
32
1. Obliczamy VN-objętość N-wymiarowej kuli
R =(2mE)1/2 -promień N-wymiarowej kuli C-stała zależna o wymiaru N granice całkowania
1/2 -promień N-wymiarowej kuli. C-stała zależna o wymiaru N. granice całkowania.")
33
3. Obliczamy temperaturę
2. Obliczamy entropię: a następnie wiedząc, że 3. Obliczamy temperaturę lub wiedząc, że 4. Obliczamy ciśnienie 5. I w ten sposób otrzymujemy równanie stanu f(p,V,T,N)=0: gdy jednostką ilości jest mol: gdzie n=N/NA R=kNA NA – liczba Avogadro
=0: gdy jednostką ilości jest mol: gdzie n=N/NA R=kNA NA – liczba Avogadro.")
34
Przykład II Obliczyć entropię układu N- oscylatorów harmonicznych jednowymiarowych. Energia całkowita układu wynosi E. A. Podejście kwantowe Energia pojedynczego oscylatora j = 1, 2, 3, ...N; nj = 0, 1, 2, ... Energia układu N oscylatorów: gdzie E0 = N0 M - suma liczb kwantowych wszystkich oscylatorów, określa całkowitą energię układu. M charakteryzuje stan makroskopowy układu. Stan mikroskopowy – podajemy wszystkie liczby kwantowe oscylatorów. Stan makroskopowy – podajemy jedną liczbę M - może być realizowany na wiele sposobów.

35
Ilość kombinacji z powtórzeniami (M–elementów spośród N-elementów).
4 ... 3 1 2 Ilość kombinacji z powtórzeniami (M–elementów spośród N-elementów). Znalezienie prawdopodobieństwa termodynamicznego sprowadza się do znalezienia liczby sposobów rozdzielenia M identycznych „porcji h0” pomiędzy N rozróżnialnych „oscylatorów”.
. Znalezienie prawdopodobieństwa termodynamicznego sprowadza się do znalezienia liczby sposobów rozdzielenia M identycznych „porcji h0 pomiędzy N rozróżnialnych „oscylatorów .")
36
Entropia N oscylatorów jednowymiarowych o energii całkowitej E:
Temperatura : T Energia:

37
B. Podejście klasyczne:
Klasyczna energia układu N oscylatorów harmonicznych jednowymiarowych: Objętość przestrzeni fazowej (E):
:")
38
Entropia: Temperatura: E Energia: T

39
ROZKŁAD MAXWELLA-BOLTZMANNA - wyprowadzenie
(x,y,z,px,py,pz) py pz px c z a y x b x y N odróżnialnych cząstek w izolowanym naczyniu o objętości V=abc N punktów w przestrzeni fazowej Z izolacji układu wynika, że: całkowita liczba cząstek w układzie N=const całkowita energia układu E=const
 py. pz. px. c. z. a. y. x. b. x. y. N odróżnialnych cząstek w izolowanym naczyniu o objętości V=abc. N punktów w przestrzeni fazowej Z izolacji układu wynika, że: całkowita liczba cząstek w układzie N=const. całkowita energia układu E=const.")
40
Energia cząstki w i-tej komórce:
i – indeks komórki lub poziomu energetycznego Z warunku zachowania liczby cząstek (masy) wynika, że: Z warunku zachowania energii wynika, że:
 wynika, że: Z warunku zachowania energii wynika, że:")
41
Gdy N jest bardzo duże (~ NA) liczba różnych rozmieszczeń cząstek pomiędzy komórki przestrzeni fazowej (lub poziomy energetyczne) nie naruszających warunku zachowania liczby cząstek i energii układu jest bardzo duża. Gdy cząstki są odróżnialne, dane liczbowe rozmieszczenie pomiędzy komórki lub poziomy energetyczne można zrealizować na sposobów. N1, N2, N3, N4, N5, N6 1, 2, 2, 2, 2, 1 N1, N2, N3, N4, N5, N6 3, 2, 2, 0, 1, 2 N1, N2, N3, N4, N5, N6 4, 2, 0, 1, 0, 3

42
Najbardziej prawdopodobny jest ten rozkład, który jest realizowany na największą liczbę sposobów W. Aby ten rozkład znaleźć musimy znaleźć maksimum funkcji W. Warunek na ekstremum (maksimum): Zmienne ni nie są niezależne, ponieważ muszą spełniać omówione wcześniej warunki. By wyznaczyć ekstremum musimy zastosować metodę nieoznaczonych mnożników Lagrange’a.
: Zmienne ni nie są niezależne, ponieważ muszą spełniać omówione wcześniej warunki. By wyznaczyć ekstremum musimy zastosować metodę nieoznaczonych mnożników Lagrange’a.")
43
Sumując stronami dostajemy:
Prawdopodobieństwo, że cząstka ma energię k. k (1,3810-23 J/K) – stała Boltzmanna T(K) – temperatura absolutna
 – stała Boltzmanna. T(K) – temperatura absolutna.")
44
Przejście od rozkładu dyskretnego do ciągłego:
Obliczanie średniej energii kinetycznej cząstki o masie m i wyznaczanie

45
Obliczanie średniej energii kinetycznej cząstki o masie m i wyznaczanie c.d.

46
Przykłady obliczania średnich i wariancji (odchyleń standardowych)
Niech będzie dana wielkość mechaniczna M, która jest funkcją pędów i współrzędnych. Średnią wartość M obliczymy ze wzoru:

47
Średnia wartość Średnia wartość Średnia wartość Średnia wartość

48
Rozkład energii kinetycznej cząstki poruszającej się w 3 wymiarach :

49
Gaz doskonały w polu zewnętrznym
energia potencjalna cząsteczki w polu zewnętrznym energia ruchu postępowego, energia wewnętrzna energia całkowita funkcja Hamiltona Gęstość gazu, czyli liczba cząsteczek w jednostce objętości.

50
n0= gęstość przy zerowym polu potencjału
x,y,z – współrzędne środka masy cząsteczki Rozkład Boltzmanna: rozkład cząsteczek gazu doskonałego w polu sił zewnętrznych (w polu potencjału u(x,y,z))
)")
51
Pole sił ciężkości Gęstość cząsteczek na wysokości h nad poziomem morza. Równanie stanu gazu doskonałego F(p,V,T,N)=0 Zależność ciśnienia gazu od wysokości i masy

52
ZESPÓŁ KANONICZNY T,V,N – const. (zespół układów zamkniętych)
Jakie jest prawdopodobieństwo, że przypadkowo wybrany układ zespołu ma energię Ei? wymiana energii stała liczba układów w zespole stała całkowita energia zespołu T,V,N Ei – dozwolone energie układu Li - liczba układów o energii Ei w zespole izolacja Liczba sposobów realizacji rozkładu układów zespołu pomiędzy dozwolone poziomy energetyczne {Li}.

53
E6 E6 E6 E5 E5 E5 E4 E4 E4 E3 E3 E3 E2 E2 E2 E1 E1 E1 L1, L2, L3, L4, L5, L6 3, 2, 2, 0, 1, 2 L1, L2, L3, L4, L5, L6 2, 2, 2, 1, 1, 2 L1, L2, L3, L4, L5, L6 4, 2, 0, 1, 0, 3 W

54
Zmienne Li nie są niezależne !!!
Gdy Li jest bardzo duże istnieje jeden rozkład, dla którego W jest dużo większe niż dla wszystkich pozostałych rozkładów (W ma ostre maksimum). Szukamy tego rozkładu!!! Prawdopodobieństwo realizacji określonego rozkładu W warunek na ekstremum (maksimum) rozkład Zmienne Li nie są niezależne !!! Po pomnożeniu drugiej równości przez stały mnożnik , trzeciej przez mnożnik i dodaniu do siebie stronami tych trzech równości otrzymamy
. Szukamy tego rozkładu!!! Prawdopodobieństwo realizacji określonego rozkładu. W. warunek na ekstremum (maksimum) rozkład. Zmienne Li nie są niezależne !!! Po pomnożeniu drugiej równości przez stały mnożnik , trzeciej przez mnożnik i dodaniu do siebie stronami tych trzech równości otrzymamy.")
55
*******************************************************************************************
+ + niezależne

56
Wyznaczanie wartości mnożnika
Aby skorzystać z tej zależności, należy wyznaczyć wartości nieoznaczonych mnożników Lagrange’a. Liczba układów o energii Ei. Wyznaczanie wartości mnożnika sumowanie po stanach układu Prawdopodobieństwo tego, że dowolnie wybrany układ zespołu ma energię Ei.

57
Prawdopodobieństwo tego, że dowolnie wybrany układ zespołu ma energię Ei.
Kanoniczna suma stanów. Średnia energia wewnętrzna układu: Wyznaczanie wartości mnożnika Znajdziemy kanoniczną sumę stanów dla układu składającego się z N cząsteczek gazu doskonałego posiadających jedynie translacyjne stopnie swobody . Obliczymy średnią energię układu z powyższego wzoru i porównamy ją ze średnią energią kinetyczną gazu wynikającą z zasady ekwipartycji energii.

58
Faktoryzacja (rozkład na czynniki) kanonicznej sumy stanów
B A Gdy energia układu jest sumą energii podukładów to kanoniczna suma stanów układu daje się zapisać jako iloczyn kanonicznych sum stanów podukładów. Gdy energia układu daje się zapisać jako suma wkładów związanych z poszczególnymi stopniami swobody to

59
Obliczanie Q dla N cząsteczek gazu doskonałego (niezależnych):
Jeżeli cząsteczki są niezależne (nie oddziałują między sobą) to całkowita energia jest sumą energii poszczególnych cząsteczek: - energia pojedynczej cząsteczki n – numer cząsteczki W takim przypadku kanoniczna suma stanów układu da się wyrazić jako: Kanoniczna suma stanów dla jednej cząsteczki – cząsteczkowa suma stanów. Czynnik N! uwzględnia nieodróżnialność identycznych cząsteczek. Średnią energię cząsteczki obliczymy ze wzoru:
 to całkowita energia jest sumą energii poszczególnych cząsteczek: - energia pojedynczej cząsteczki. n – numer cząsteczki. W takim przypadku kanoniczna suma stanów układu da się wyrazić jako: Kanoniczna suma stanów dla jednej cząsteczki – cząsteczkowa suma stanów. Czynnik N! uwzględnia nieodróżnialność identycznych cząsteczek. Średnią energię cząsteczki obliczymy ze wzoru:")
60
Obliczanie cząsteczkowej sumy stanów:
Dozwolone wartości energii cząsteczki poruszającej się w sześciennym naczyniu o objętości V =L3 wynoszą: m – masa cząsteczki L – wymiar sześcianu nx,ny,nz = 1,2,3,..., - liczby kwantowe Cząsteczkowa suma stanów ma postać: Możemy ją zapisać jako iloczyn trzech identycznych sum:

61
Ponieważ A jest bardzo małe zastępujemy sumowanie całkowaniem.
Obliczamy średnią energię: Z porównania: dostaniemy:

62
Średnia energia układu - energia wewnętrzna:
Zmiana energii wewnętrznej: Ei – możliwa energia układu Pi – prawdopodobieństwo, że układ ma energię Ei - 1/kT Q – kanoniczna suma stanów V – objętość układu

63
-p I zasada termodynamiki: dE=Q+WdE=TdS-pdV

64
Związki kanonicznej sumy stanów z funkcjami termodynamicznymi układu
p H - - U G Potencjał termodynamiczny, czyli funkcja która w stanie równowagi układu zamkniętego osiąga minimum. + + F V T

65
ZESPÓŁ KANONICZNY T,V,N – const. - rozkład klasyczny
Rozważmy układ zamknięty zanurzony w otoczeniu o temperaturze T. Załóżmy, że: Rozmiar układu jest bardzo mały w porównaniu z rozmiarem otoczenia. Energia wymieniana przez układ z otoczeniem stanowi znikomą część energii układu. Stan otoczenia nie ulega zmianie. Niech układ znajduje się w stanie o energii Hu. Liczba mikrostanów całości wyniesie (H-Hu). Ponieważ Hu <<H, rozwijamy (H-Hu) w szereg wokół Hu=0 . T,V,N
. Ponieważ Hu <<H, rozwijamy (H-Hu) w szereg wokół Hu=0 . T,V,N.")
66
Z warunku unormowania wynika, że:
H – hamiltonian F – liczba stopni swobody Q-1 – stała normująca Kanoniczna suma stanów dla układu klasycznego. Kanoniczna suma stanów dla układu kwantowego. i – stany k- poziomy energetyczne gk – stopień degeneracji poziomu o energii Ek

67
PODSUMOWANIE Kanoniczna suma stanów : Kwantowo:
Ek - poziomy energetyczne układu gk - stopień degeneracji k-tego poziomu Klasycznie: H(p,q) - hamiltonian układu F – liczba stopni swobody układu = Nf
 - hamiltonian układu F – liczba stopni swobody układu = Nf.")
68
Kanoniczna suma stanów dla N cząsteczek gazu doskonałego w objętości V:
q(V,T) – cząsteczkowa suma stanów (suma stanów dla jednej cząsteczki) N! – liczba permutacji N identycznych cząsteczek Cząsteczkową sumę stanów obliczamy ze wzorów: kwantowo klasycznie
 – cząsteczkowa suma stanów (suma stanów dla jednej cząsteczki) N! – liczba permutacji N identycznych cząsteczek. Cząsteczkową sumę stanów obliczamy ze wzorów: kwantowo klasycznie.")
69
Związki kanonicznej sumy stanów i cząsteczkowej sumy stanów z funkcjami termodynamicznymi:
Energia swobodna F = potencjał termodynamiczny, czyli funkcja termodynamiczna, która w stanie równowagi osiąga minimum. Energia wewnętrzna U=E Entropia S Ciśnienie p Potencjał chemiczny

70
Obliczenie całki statystycznej dla rzeczywistych układów jest trudne:
trudności matematyczne: całka statystyczna dla układów makroskopowych ma bardzo dużą krotność (dla 1 mola: ~1024 krotna); trudności fizyczne: ograniczone informacje o oddziaływaniach w rzeczywistych układach; z reguły nie znamy poziomów energetycznych kwantowych układów makroskopowych i stopni ich degeneracji. Posługujemy się układami modelowymi.
; trudności fizyczne: ograniczone informacje o oddziaływaniach w rzeczywistych układach; z reguły nie znamy poziomów energetycznych kwantowych układów makroskopowych i stopni ich degeneracji. Posługujemy się układami modelowymi.")
71
OBLICZANIE CZĄSTECZKOWEJ SUMY STANÓW DLA GAZU ATOMOWEGO I DWUATOMOWEGO
Gaz atomowy: - translacyjne stopnie swobody x,y,z - wewnętrzne stopnie swobody – stany elektronowe energia atomu suma stanów dla atomu Gdy to

72
Różnice energii pierwszego wzbudzonego i podstawowego stanu elektronowego
Cl N2 K 1 300 K K Jeśli za zero energii przyjmiemy 0 to: wkład translacyjny elektronowy jądrowy g0 - stopień degeneracji podstawowego poziomu energii elektronowej =2s+1 - stopień degeneracji spinowej jądra (s-liczba spinowa jądra)
")
73
Funkcje termodynamiczne doskonałego gazu atomowego
Energia swobodna Entropia Potencjał chemiczny Potencjał standardowy

74
Równanie stanu gazu doskonałego
Ogólna postać równania stanu: Energia swobodna gazu doskonałego: Ciśnienie gazu doskonałego:

75
Entropia molowa Zmiana entropii - w stałej objętości
- w stałej temperaturze - w stałym ciśnieniu

76
Pojemność cieplna Pojemność cieplna - stosunek ilości ciepła (dQ) dostarczonego do układu do odpowiadającego mu przyrostu temperatury (dT). gdzie: C - pojemność cieplna Q - energia cieplna T - temperatura Pojemność cieplna przypadająca na jednostkę masy to ciepło właściwe, a na 1 mol to ciepło molowe.

77
Energia wewnętrzna (kinetyczna) gazu atomowego.
Suma stanów dla atomu. Energia wewnętrzna (kinetyczna) gazu atomowego. Molowa pojemność cieplna atomowego gazu doskonałego.
 gazu atomowego. Molowa pojemność cieplna atomowego gazu doskonałego.")
78
Cząsteczki dwuatomowe - wkład rotacyjny i oscylacyjny
Wkład rotacji do kanonicznej sumy stanów cząsteczki dwuatomowej Jeśli ruch rotacyjny opisujemy modelem rotatora sztywnego kwantowego to: J = 0,1,2,.... stopień degeneracji J – tego poziomu energetycznego Wkład rotacji do cząsteczkowej sumy stanów wyniesie: charakterystyczna temperatura rotacji Dla wysokich temperatur, , sumowanie możemy zastąpić całkowaniem - liczba symetrii równa 1 dla cząsteczek heterojądrowych i 2 dla cząsteczek homojądrowych

79
Wkład rotacji do pojemności cieplnej gazu dwuatomowego:
rotacyjna suma stanów (wysokie temperatury) dla cząsteczki dla N cząsteczek Energia rotacji
 dla cząsteczki. dla N cząsteczek. Energia rotacji.")
80
Wkład oscylacji do cząsteczkowej sumy stanów
Drgania w cząsteczce opisujemy modelem oscylatora harmonicznego. cząsteczka dwuatomowa – oscylator harmoniczny jednowymiarowy cząsteczka n atomowa – 3n-5 (cząsteczka liniowa) lub 3n-6 (cząsteczka nieliniowa) oscylacji harmonicznych jednowymiarowych Ad.1 Energia jednowymiarowego oscylatora harmonicznego dana jest wzorem: - mech. kwantowa = 0,1, g =1 Cząsteczkowa oscylacyjna suma stanów: Szereg geometryczny z ilorazem q << 1, czyli zbieżny. charakterystyczna temperatura oscylacji
 lub 3n-6 (cząsteczka. nieliniowa) oscylacji harmonicznych jednowymiarowych. Ad.1 Energia jednowymiarowego oscylatora harmonicznego dana jest wzorem: - mech. kwantowa = 0,1,2 g =1. Cząsteczkowa oscylacyjna suma stanów: Szereg geometryczny z ilorazem q << 1, czyli zbieżny. charakterystyczna temperatura oscylacji.")
81
Wkład oscylacji do pojemności cieplnej
Wkład oscylacji do pojemności cieplnej gazu dwuatomowego: Oscylacyjna suma stanów dla cząsteczki dla N cząsteczek Energia oscylacji Wkład oscylacji do pojemności cieplnej

82
Tabela: Charakterystyczne temperatury rotacji niektórych cząsteczek
H D N2 O HCl HI rot ,85 2, , ,0 Tabela: Charakterystyczne temperatury oscylacji niektórych cząsteczek H2 Cl2 I2 N2 O2 HCl CO osc Wkład rotacji do pojemności cieplnej Pojemność cieplna gazu dwuatomowego

83
Kwantowanie energii poszczególnych rodzajów ruchów:
- ruch translacyjny w nieskończonej objętości nie jest kwantowany a dozwolone poziomy energetyczne tworzą kontinuum, - ruch rotacyjny jest kwantowany a odległości pomiędzy poziomami energetycznymi zależą od momentu bezwładności cząsteczki i rosną ze wzrostem stanu wzbudzenia, - ruch oscylacyjny jest kwantowany, odległości pomiędzy poziomami energetycznymi zależą od mas atomów tworzących cząsteczkę oraz energii wiązań między nimi. - elektronowe poziomy energetyczne są kwantowane a typowe odległości między nimi są bardzo duże.

84
T < T2 Obsadzenie poziomów energetycznych cząsteczki maleje wykładniczo ze wzrostem energii. W bardzo wysokiej temperaturze wszystkie poziomy są obsadzone mniej więcej równomiernie.

85
Rozkłady Boltzmanna dla trzech rodzajów ruchu w tej samej temperaturze
Rozkłady Boltzmanna dla trzech rodzajów ruchu w tej samej temperaturze. Skale energii dla każdego rodzaju ruchu są różne. C 3R/2 Cpost. granica klasyczna R Cosc. Crot. T

86
Wielki zespół kanoniczny – zespół grandkanoniczny
Zespół układów otwartych – układy wymieniają z otoczeniem energię E i cząstki N (zmiany energii i liczby cząstek mają charakter fluktuacji). Objętość układu jest stała oraz stałe są pewne parametry zewnętrzne narzucone przez otoczenie (T,). Otoczeniem jest nieskończenie duży zbiornik energii i cząstek dla danego układu. Oddziaływanie z otoczeniem jest na tyle słabe, że układy zespołu można uważać za quasi-niezależne statystycznie. Zespół składa się z układów zawierających wszystkie możliwe liczby cząstek od 0 do pewnej maksymalnej wartości NL i różnej energii. L – liczba układów w zespole LNi - liczba układów zespołu zawierających w danej chwili czasu po N cząstek każdy i znajdujących się w i-tym stanie energetycznym. Ei(N,V) – energia układu zawierającego N cząstek w objętości V . Wymiana cząstek i energii między układami stała liczba układów w zespole stała całkowita liczba cząstek w zespole stała całkowita energia zespołu T,V, izolacja
. Objętość układu jest stała oraz stałe są pewne parametry zewnętrzne narzucone przez otoczenie (T,). Otoczeniem jest nieskończenie duży zbiornik energii i cząstek dla danego układu. Oddziaływanie z otoczeniem jest na tyle słabe, że układy zespołu można uważać. za quasi-niezależne statystycznie. Zespół składa się z układów zawierających wszystkie możliwe liczby cząstek. od 0 do pewnej maksymalnej wartości NL i różnej energii. L – liczba układów w zespole. LNi - liczba układów zespołu zawierających w danej chwili czasu po N cząstek. każdy i znajdujących się w i-tym stanie energetycznym. Ei(N,V) – energia układu zawierającego N cząstek w objętości V. . Wymiana cząstek i energii między układami. stała liczba układów w zespole stała całkowita liczba cząstek w zespole stała całkowita energia zespołu. T,V, izolacja.")
87
Jakie jest prawdopodobieństwo, że przypadkowo wybrany układ zespołu zawiera N cząstek i ma energię E Ni? zależne W celu znalezienia maksimum lnW stosujemy metodę nieoznaczonych mnożników Lagrange’a. + + 87 87 +

88
niezależne Prawdopodobieństwo tego, że układ otwarty ma N cząsteczek i energię ENi. Wielka kanoniczna suma stanów. Można wykazać, że : - potencjał chemiczny - aktywność absolutna

89
Obliczanie funkcji termodynamicznych układu otwartego przy pomocy wielkiej sumy stanów:
Potencjał termodynamiczny J osiąga minimum w stanie równowagi układu otwartego. Średnia energia układu: Średnia liczba cząsteczek w układzie:

90
Entalpia swobodna układu:
Energia swobodna układu: Entropia układu: Ciśnienie: Przestrzeń fazowa dla N cząstek ma wymiar: 2fN. Ponieważ w układach otwartych liczba cząstek ulega zmianie, to stanu wielkiego zespołu kanonicznego nie można przedstawić w jednej przestrzeni fazowej. Różnym wartościom N odpowiadają przestrzenie fazowe różnych wymiarów. Musimy mieć zbiór przestrzeni fazowych.

91
Fluktuacje Fluktuacje - to przypadkowe odchylenia wielkości fizycznych od ich wartości średnich. Dla wielkości addytywnych, tj. proporcjonalnych do ilości cząstek układu N, dyspersja danej wielkości A(t) związana z jej fluktuacjami jest proporcjonalna do N. Wobec tego względna fluktuacja σ(A) / A jest proporcjonalna do (N)-1/2. Stąd dla układów makroskopowych, gdzie N jest rzędu liczby Avogadro, wpływ fluktuacji jest poza zasięgiem pomiaru. W układach o małej liczbie cząstek względne fluktuacje mogą być bardzo duże. Dlatego znajomość wartości średnich nie wystarcza do opisu zachowania się małego układu.
 związana z jej fluktuacjami. jest proporcjonalna do N. Wobec tego względna fluktuacja σ(A) / A jest proporcjonalna do (N)-1/2. Stąd dla układów makroskopowych, gdzie N jest rzędu liczby Avogadro, wpływ fluktuacji jest poza zasięgiem pomiaru. W układach o małej liczbie cząstek względne fluktuacje mogą być bardzo duże. Dlatego znajomość wartości średnich nie wystarcza do opisu zachowania się małego układu.")
92
Fluktuacje wielkości fizycznych (np
Fluktuacje wielkości fizycznych (np. gęstości, temperatury, prądu elektrycznego) są wynikiem atomowej struktury materii. Fluktuacje termodynamiczne są wywołane ruchami cieplnymi, fluktuacje kwantowe są związane z kwantowymi relacjami nieokreśloności (zasada nieokreśloności Heisenberga). Z fluktuacjami związanych jest wiele zjawisk, np. : Ruchy Browna. Ruch cząstki jest rezultatem tego, że zderzenia z cząsteczkami roztworu nie kompensują się całkowicie w każdej chwili czasu, chociaż średnia czasowa działających sił jest równa zeru. Fluktuacje gęstości atmosfery powodują różne rozpraszanie światła (zmiana odcienia błękitu nieba, opalescencja). Fluktuacje ograniczają czułość przyrządów pomiarowych, powodują zmiany położenia wskazówek, szumy wzmacniaczy itp.., czyli fałszują wyniki pomiarów.
 są wynikiem atomowej struktury materii. Fluktuacje termodynamiczne są wywołane ruchami cieplnymi, fluktuacje kwantowe są związane z kwantowymi relacjami nieokreśloności (zasada nieokreśloności Heisenberga). Z fluktuacjami związanych jest wiele zjawisk, np. : Ruchy Browna. Ruch cząstki jest rezultatem tego, że zderzenia z cząsteczkami roztworu nie kompensują się całkowicie w każdej chwili czasu, chociaż średnia czasowa działających sił jest równa zeru. Fluktuacje gęstości atmosfery powodują różne rozpraszanie światła (zmiana odcienia błękitu nieba, opalescencja). Fluktuacje ograniczają czułość przyrządów pomiarowych, powodują zmiany położenia wskazówek, szumy wzmacniaczy itp.., czyli fałszują wyniki pomiarów.")
93
Rodzaje fluktuacji: lokalne fluktuacje wewnętrzne (np. lokalne fluktuacje gęstości przy stałej objętości całkowitej układu i stałej całkowitej liczbie cząstek); fluktuacje parametrów termodynamicznych dla układu jako całości (mogą zachodzić dla parametrów, które nie są ustalone przez warunki izolacji: zatem dla układu izolowanego są możliwe tylko fluktuacje lokalne. Rozważając istnienie lokalnych fluktuacji możemy wyprowadzić wzór na fluktuację dowolnej wielkości X. Załóżmy, że mamy bardzo duży układ zamknięty a w nim bardzo mały układ, w którym różne parametry mogą odchylać się o pewną wartość. Dla dowolnego parametru X będącego funkcją X = X(T, V) lub X = X(S, p) możemy zapisać następujące zależności:
; fluktuacje parametrów termodynamicznych dla układu jako całości (mogą zachodzić dla parametrów, które nie są ustalone przez warunki izolacji: zatem dla układu izolowanego są możliwe tylko fluktuacje lokalne. Rozważając istnienie lokalnych fluktuacji możemy wyprowadzić wzór na fluktuację dowolnej wielkości X. Załóżmy, że mamy bardzo duży układ zamknięty a w nim bardzo mały układ, w którym różne parametry mogą odchylać się o pewną wartość. Dla dowolnego parametru X będącego funkcją X = X(T, V) lub X = X(S, p) możemy zapisać następujące zależności:")
94
Wykazano przy tym, że rozkład fluktuacji dla zmiennych S, T, V, p jest rozkładem Gaussa:
gdzie y= S, T, V, p Ponadto wykazano, że

95
Fluktuacje energii w zespole kanonicznym
Średnia fluktuacja kwadratowa energii - wariancja Dla atomowego gazu doskonałego: Fluktuacja względna: Funkcja rozkładu energii dla układu zespołu kanonicznego

96
Fluktuacje liczby cząstek w wielkim zespole kanonicznym
Wariancja liczby cząstek w układzie otwartym: Dla gazu doskonałego: Fluktuacje liczby cząstek są zwykle zaniedbywalne. W gazach rozrzedzonych, mogą być znaczne, np. w górnej warstwie atmosfery - błękitny kolor nieba.

97
òò å gk å å Równoważność zespołów statystycznych dpdq e N! h 1 lub =
Zespół mikrokanoniczny (E,V,N - wszystkie ekstensywne i mechaniczne) S=kln(E,V,N) Zespół kanoniczny (N,V,T - dwa ekstensywne i mechaniczne i jeden termiczny i intensywny) F=-kTlnQ(T,V,N) dpdq e N! h 1 Q lub ) q , p ( H fN k E i òò å gk å b - = Wielki zespół kanoniczny (V,T, - jeden mechaniczny i ekstensywny, dwa termiczne i intensywne) J=-pV=-kTln(V,T,) å m b + - l = X N i ENi Q e
 S=kln(E,V,N) Zespół kanoniczny (N,V,T - dwa ekstensywne i mechaniczne i jeden termiczny i intensywny) F=-kTlnQ(T,V,N) dpdq. e. N! h. 1. Q. lub. ) q. , p. ( H. fN. k. E. i. òò. å gk. å. b. - = Wielki zespół kanoniczny (V,T, - jeden mechaniczny i ekstensywny, dwa termiczne i intensywne) J=-pV=-kTln(V,T,) å. m. b. + - l. = X. N. i. ENi. Q. e.")
98
pdV=0 SdT dF - = dV=0 T p dU 1 dS - = m =0 = Nd pdV SdT ) pV ( d +
Potencjały termodynamiczne - to funkcje, które w stanie równowagi w zadanych warunkach osiągają ekstremum dV=0 T p dU 1 dS - = Zespół mikrokanoniczny W układzie izolowanym w stanie równowagi entropia osiąga maksimum. pdV=0 SdT dF - = Zespół kanoniczny W układzie zamkniętym w stanie równowagi energia swobodna osiąga minimum. Wielki zespół kanoniczny W układzie otwartym w stanie równowagi potencjał termodynamiczny J = - pV osiąga minimum. m =0 + = Nd pdV SdT ) pV ( d
 pV. ( d.")
99
å Czy różne zespoły dają wyniki zgodne ze sobą? l = X ) N , V T ( Q l
Przejście od jednego rozkładu do drugiego jest możliwe metodą członu maksymalnego. Dla układów makroskopowych człony tej sumy przechodzą przez niezwykle ostre maksimum, możliwe jest więc zastąpienie sumy członem maksymalnym. å l = X N Q ) N , V T ( Q * l = X * N Q ln + l = X - jest rzędu N* Przypuśćmy, że w sumie mamy m członów porównywalnych z *, tzn. = m *. Wtedy ln =ln * +ln(m) Gdy N*=1020 i m=N* to ln(m) = 46<<N*
 N. , V. T. ( Q. * l. = X. * N. Q. ln. + l. = X. - jest rzędu N* Przypuśćmy, że w sumie mamy m członów porównywalnych z *, tzn. = m *. Wtedy. ln =ln * +ln(m) Gdy N*=1020 i m=N* to ln(m) = 46<<N*")
100
N Q ln = ¶ + l Þ F ln b - = Q Z warunku na ekstremum otrzymujemy:
N Q ln = + l Þ Pamiętając, że oraz F Q ln N b - = otrzymamy: Jest to zależność termodynamiczna słuszna gdy N*= Podobnie możemy w kanonicznej sumie stanów wyznaczyć człon maksymalny:

101
= ln k S g(E,V,N) Z warunku na ekstremum otrzymujemy: Jeżeli
to z powyższego równania wynika, że Zatem wszystkie trzy rozkłady są równoważne z termodynamicznego punktu widzenia. Różnice między nimi polegają na fluktuacjach.

102
Schemat obliczeń dla zespołu kanonicznego
Obliczamy całkę statystyczną Q dla zespołu kanonicznego. Obliczamy energię swobodną: F = -kT lnQ. Obliczamy pozostałe funkcje termodynamiczne. Ustalamy związki pomiędzy parametrami układu Uwaga: w warunkach równowagi (V,T=const) energia swobodna F osiąga minimum. Schemat obliczeń dla wielkiego zespołu statystycznego: Obliczamy wielką sumę statystyczną ; Obliczamy potencjał termodynamiczny Obliczamy pozostałe funkcje termodynamiczne przy użyciu J; Ustalamy związki między parametrami układu. # Korzysta się często z równania na średnią liczbę cząstek układu oraz warunku równowagi chemicznej z otoczeniem: .
 energia swobodna F osiąga minimum. Schemat obliczeń dla wielkiego zespołu statystycznego: Obliczamy wielką sumę statystyczną ; Obliczamy potencjał termodynamiczny. Obliczamy pozostałe funkcje termodynamiczne przy użyciu J; Ustalamy związki między parametrami układu. # Korzysta się często z równania na średnią liczbę cząstek układu oraz warunku równowagi chemicznej z otoczeniem: .")
103
Przykłady zastosowań:
Równanie stanu jest związkiem między parametrami układu termodynamicznego takimi jak ciśnienie p, gęstość ρ (N/V), temperatura T, entropia s (S/n), energia wewnętrzna u (U/n), który można zapisać w postaci następujących równań: Dla gazu doskonałego równanie stanu ma postać : Wykaż jego słuszność posługując się zespołem kanonicznym.
, temperatura T, entropia s (S/n), energia wewnętrzna u (U/n), który można zapisać w postaci następujących równań: Dla gazu doskonałego równanie stanu ma postać : Wykaż jego słuszność posługując się zespołem kanonicznym.")
104
Przykład 1 c.d. T, V, N = const. Gdy brak jest oddziaływań międzycząsteczkowych: - energia pojedynczej cząsteczki n – numer cząsteczki W takim przypadku kanoniczna suma stanów układu da się wyrazić jako: Sumowanie po poziomach energetycznych cząsteczki. lub Całkowanie po pędach i współrzędnych pojedynczej cząsteczki. Czynnik uwzględniający nieodróżnialność identycznych cząsteczek.

105
Przykład 1 c.d. Gdy cząsteczka wykonuje jedynie ruch postępowy (np. atom), to jej energia wyraża się wzorem: kwantowo: klasycznie: Cząsteczkowa suma stanów jest proporcjonalna do objętości, w której cząsteczka się porusza. Obliczamy ciśnienie: Równanie stanu gazu doskonałego.

106
Przykład 2 W naczyniu o objętości V znajduje się mieszanina dwóch gazów doskonałych A i B. Wykazać, że ciśnienie mieszaniny jest równe sumie ciśnień cząstkowych gazów: PRAWO DALTONA E = EA + EB energia układu Q = QAQB kanoniczna suma stanów układu V, T, NA, NB QA i QB – kanoniczne sumy stanów pojedynczych gazów A i B zajmujących objętość V

107
Przykład 2 c.d. Obliczamy swobodną energię układu: Obliczamy ciśnienie w układzie: c.d.n.

108
Przykład 3 Naczynie z gazem przedzielono ruchomą przegrodą na dwie części A i B. Przegroda przemieszcza się dopóki układ nie osiągnie stanu równowagi. Kiedy to nastąpi? T=const., NA=const., NB=const. VA+VB=V=const. dVA = - dVB VA i VB zmienne bo przegroda jest ruchoma W równowagowym położeniu przegrody energia swobodna układu osiąga minimum, czyli dF = 0 T, VA, NA T, VB, NB (A) (B) W stanie równowagi ciśnienia po obu stronach bariery są równe.
 (B) W stanie równowagi ciśnienia po obu stronach bariery są równe.")
109
Przykład 3 c.d. Naczynie z gazem przedzielono nieruchomą przegrodą z otworem na dwie części A i B. Cząsteczki wędrują z jednej części do drugiej dopóki układ nie osiągnie stanu równowagi. Kiedy to nastąpi? T=const., VA=const., VB=const. NA+NB=N=const. dNB = - dNA NA i NB zmienne bo przegroda ma otwór. W stanie równowagi energia swobodna układu osiąga minimum, czyli dF = 0 T, VA, NA T, VB, NB (A) (B) W stanie równowagi potencjały chemiczne gazu po obu stronach bariery są równe.
 (B) W stanie równowagi potencjały chemiczne gazu po obu stronach bariery są równe.")
110
Przykład 4 Wyprowadzenie równania na stałą równowagi reakcji odwracalnej zachodzącej w doskonałej fazie gazowej: Warunek równowagi dF(T,V,NA, NB, NC, ND)=0 lub bo Liczby cząsteczek substratów i produktów mogą się zmieniać zgodnie ze stechiometrycznym zapisem reakcji. Stosunek przyrostów liczby cząsteczek do współczynników stechiometrycznych dla substratów i produktów reakcji musi być taki sam. - liczba postępu reakcji Zmianę liczby cząsteczek każdego składnika w układzie możemy wyrazić poprzez zmianę .
=0. lub. bo. Liczby cząsteczek substratów i produktów mogą się zmieniać zgodnie ze stechiometrycznym zapisem reakcji. Stosunek przyrostów liczby cząsteczek do współczynników stechiometrycznych dla substratów i produktów reakcji musi być taki sam. - liczba postępu reakcji. Zmianę liczby cząsteczek każdego składnika w układzie możemy wyrazić poprzez zmianę .")
111
ogólny warunek równowagi
Dla mieszaniny gazów doskonałych (A+B+C+D): F = -kTlnQ Q=QA QB QC QD kanoniczna suma stanów qA – cząsteczkowa (molekularna) funkcja rozdziału Energia swobodna układu jest sumą energii dla poszczególnych składników.
: F = -kTlnQ. Q=QA QB QC QD. kanoniczna suma stanów. qA – cząsteczkowa (molekularna) funkcja rozdziału. Energia swobodna układu jest sumą energii dla poszczególnych składników.")
112
Wstawiamy wyrażenia na potencjały do ogólnego warunku równowagi:
Wyrażenie w nawiasie zwijamy pod jeden logarytm: lub Logarytm jest równy 0, gdy liczba logarytmowana jest równa jedności: Z powyższej zależności wynika, że jeżeli stężenia składników mieszaniny reakcyjnej będziemy określać liczbą cząsteczek, to stała równowagi przyjmie postać: lub

113
MODELE SIATKOWE Model Isinga
(Model stosowany głównie w teoriach ferromagnetyków i stopów dwuskładnikowych, ale jego znaczenie jest znacznie ogólniejsze)
")
114
Z ferromagnetyzmem mamy do czynienia wtedy, gdy zbiór spinów atomowych ulega uporządkowaniu w ten sposób, że momenty magnetyczne z nimi związane skierowane są w tym samym kierunku, dając niezerowy wypadkowy moment magnetyczny w sensie makroskopowym. Najprostszym teoretycznym modelem ferromagnetyzmu jest model Isinga. Model ten jako pierwszy zaproponował Wilhelm Lenz w 1920 roku: Nazwa modelu pochodzi od nazwiska jego studenta Ernsta Isinga, który uczynił go tematem swojej pracy doktorskiej w 1925 roku. Wyobraźmy sobie ferromagnetyk jako kryształ o idealnej sieci, w której każdy z N węzłów jest obsadzony przez atomy obdarzone spinem s=1/2. Kryształ umieszczony jest w zewnętrznym polu magnetycznym o natężeniu H. Oznaczmy możliwe dwa stany kwantowe spinów (orientacje) przez si= –1 () lub +1 (). Energia oddziaływania jest sumą oddziaływań par spinów między sobą oraz energii oddziaływania spinów z polem magnetycznym, przy czym zakładamy dodatkowo, że oddziałują jedynie najbliżsi sąsiedzi. Przyjmując odpowiednie zero energii, wzór na energię możemy zapisać w postaci: <i,j> oznacza sumowanie po parach najbliższych sąsiadów, J jest tzw. energią wymiany a atomowym momentem magnetycznym.
 przez si= –1 () lub +1 (). Energia oddziaływania jest sumą oddziaływań par spinów między sobą oraz energii oddziaływania spinów z polem magnetycznym, przy czym zakładamy dodatkowo, że oddziałują jedynie najbliżsi sąsiedzi. Przyjmując odpowiednie zero energii, wzór na energię możemy zapisać w postaci: <i,j> oznacza sumowanie po parach najbliższych sąsiadów, J jest tzw. energią wymiany a atomowym momentem magnetycznym.")
115
Powyższe równanie stanowi sformułowanie modelu Isinga
Powyższe równanie stanowi sformułowanie modelu Isinga. Pierwszy człon w tym równaniu wynika z elektrostatycznych oddziaływań między sąsiednimi atomami. Energia oddziaływań ulega obniżeniu, gdy spiny atomów są zgodne i podwyższeniu, gdy są przeciwne (zakaz Pauliego). Różne rozmieszczenie ładunków w przestrzeni daje różne energie oddziaływania, których miarą jest energia wymiany J. Ponieważ energia wymiany jest pochodzenia elektrostatycznego jej wartość może być znaczna, zwykle J ~ 1eV. Jest to wartość dużo większa niż energia związana z bezpośrednim oddziaływaniem sąsiadujących spinów atomowych, która jest rzędu 10-4 eV. Z uwagi na to, że efekt wymiany jest krótkozasięgowy, ograniczenie oddziaływań do najbliższych sąsiadów jest uzasadnione. Przeanalizujmy model Isinga w oparciu o przybliżenie średniego pola. W ramach tego przybliżenia energię i-tego atomu w sieci możemy zapisać następująco: Sumowanie biegnie po najbliższych sąsiadach węzła i, a ½ pozwala uniknąć sumowania dwukrotnie tych samych oddziaływań w wyrażeniu na energię całkowitą:
. Różne rozmieszczenie ładunków w przestrzeni daje różne energie oddziaływania, których miarą jest energia wymiany J. Ponieważ energia wymiany jest pochodzenia elektrostatycznego jej wartość może być znaczna, zwykle J ~ 1eV. Jest to wartość dużo większa niż energia związana z bezpośrednim oddziaływaniem sąsiadujących spinów atomowych, która jest rzędu 10-4 eV. Z uwagi na to, że efekt wymiany jest krótkozasięgowy, ograniczenie oddziaływań do najbliższych sąsiadów jest uzasadnione. Przeanalizujmy model Isinga w oparciu o przybliżenie średniego pola. W ramach tego przybliżenia energię i-tego atomu w sieci możemy zapisać następująco: Sumowanie biegnie po najbliższych sąsiadach węzła i, a ½ pozwala uniknąć sumowania dwukrotnie tych samych oddziaływań w wyrażeniu na energię całkowitą:")
116
Zapiszmy energię i-tego węzła w postaci:
gdzie jest efektywnym polem magnetycznym działającym na wybrany atom i jest sumą zewnętrznego pola magnetycznego i pola magnetycznego generowanego przez sąsiednie atomy. Rozważmy pojedynczy atom w polu magnetycznym Hm. Zgodnie z rozkładem Boltzmanna średnie s dla atomu wynosi: gdzie =1/kT. Utożsamiając Hm z efektywnym natężeniem pola magnetycznego możemy Heff i wyrazić następująco:

117
Zauważmy, że w tym ujęciu możemy uważać, że każdy atom znajduje się w łaźni cieplnej wytwarzanej przez pozostałe atomy. Wygodnie jest zdefiniować temperaturę krytyczną Tc i krytyczne natężenie pola magnetycznego Hc: Średnią wartość s można wtedy zapisać równaniem: Równania tego nie można rozwiązać analitycznie. Można je rozwiązać iterując wg schematu: dopóki

118
Zdefiniujmy wypadkową magnetyzację energię H=0 H=0

119
oraz pojemność cieplną
H=0 Można zauważyć, że poniżej temperatury krytycznej (temperatury Curie) występuje spontaniczna magnetyzacja, tzn. efekt wymiany jest na tyle duży aby spowodować spontaniczne porządkowanie spinów (orientację w tym samym kierunku). Z drugiej strony, termiczne fluktuacje powodują całkowity zanik uporządkowania powyżej temperatury krytycznej. Co więcej, w temperaturze krytycznej pierwsza pochodna energii po temperaturze wykazuje nieciągłość. Ta nieciągłość skutkuje gwałtownym skokiem na krzywej pojemności cieplnej gdy T/Tc =1. Raptowny zanik spontanicznej magnetyzacji po przekroczeniu temperatury krytycznej jest rodzajem przejścia fazowego.
 występuje spontaniczna magnetyzacja, tzn. efekt wymiany jest na tyle duży aby spowodować spontaniczne porządkowanie spinów (orientację w tym samym kierunku). Z drugiej strony, termiczne fluktuacje powodują całkowity zanik uporządkowania powyżej temperatury krytycznej. Co więcej, w temperaturze krytycznej pierwsza pochodna energii po temperaturze wykazuje nieciągłość. Ta nieciągłość skutkuje gwałtownym skokiem na krzywej pojemności cieplnej gdy T/Tc =1. Raptowny zanik spontanicznej magnetyzacji po przekroczeniu temperatury krytycznej jest rodzajem przejścia fazowego.")
120
Zgodnie z konwencjonalną klasyfikacją przejść fazowych przejście fazowe nazywamy przejściem fazowym pierwszego rodzaju jeśli energia jest nieciągłą funkcją parametru uporządkowania (w naszym przypadku temperatury T) i przejściem fazowym drugiego rodzaju jeśli energia jest funkcją ciągłą ale jej pierwsza pochodna względem parametru uporządkowania jest nieciągła. Zanik spontanicznego namagnesowania w ferromagnetyku powyżej temperatury krytycznej jest przejściem fazowym drugiego rodzaju.

121
Aby zaobserwować przejście fazowe pierwszego rodzaju przeanalizujmy zachowanie się magnetyzacji M jako funkcji zewnętrznego pola magnetycznego w stałej temperaturze T<Tc. H0 H0 Na rysunkach przedstawiono magnetyzację M i energię E jako funkcje natężenia zewnętrznego pola magnetycznego H w temperaturze niższej niż temperatura krytyczna (wyznaczone metodą iteracyjną). Magnetyzacja wykazuje nieciągłość charakterystyczną dla przejścia fazowego pierwszego rodzaju. Obserwujemy również stany metastabilne i histerezę w pewnym zakresie pól. Magnetyzacja w tym zakresie zależy od historii układu tzn. od tego czy H rośnie czy maleje podczas wchodzenia w obszar metastabilny.
. Magnetyzacja wykazuje nieciągłość charakterystyczną dla przejścia fazowego pierwszego rodzaju. Obserwujemy również stany metastabilne i histerezę w pewnym zakresie pól. Magnetyzacja w tym zakresie zależy od historii układu tzn. od tego czy H rośnie czy maleje podczas wchodzenia w obszar metastabilny.")
122
Na kolejnych rysunkach pokazano magnetyzację i energię jako funkcje natężenia pola magnetycznego w temperaturze równej temperaturze krytycznej (T=Tc). Magnetyzacja i energia są tym razem funkcjami ciągłymi i nie obserwujemy stanów metastabilnych. Oznacza to, że podczas zmiany pola magnetycznego przejście fazowe pierwszego rodzaju występuje jedynie poniżej temperatury krytycznej, gdy ferromagnetyk wykazuje spontaniczną magnetyzację.

123
Przybliżenie średniego pola pozwala poprawnie przewidzieć występowanie przejść fazowych pierwszego i drugiego rodzaju odpowiednio dla H0 i H=0. Szczegółowe wyniki dotyczące tych przejść nie są jednak w pełni poprawne. Aby uzyskać lepszy wynik, można zastosować metodę Monte-Carlo. Rozważmy dwuwymiarową siatkę, której węzły obsadzają atomy. Niech L oznacza wymiar siatki a N=L2 liczbę atomów w siatce jak to pokazano na poniższym rysunku:

124
Podejście Monte-Carlo do modelu Isinga, ignorujące zupełnie przybliżenie średniego pola, opiera się na następującym algorytmie: Kolejno dla każdego atomu w siatce: Oblicz zmianę energii E spowodowaną zmianą orientacji jego spinu. Jeśli E<0 zachowaj zmienioną orientację spinu. Jeśli E >0 wtedy zaakceptuj nową orientację z prawdopodobieństwem Czynności te powtarzaj do osiągnięcia stanu równowagi termicznej. Celem algorytmu jest przeczesanie wszystkich stanów w celu sprawdzenia, czy każdy z nich jest zajmowany z prawdopodobieństwem boltzmannowskim, tzn. z prawdopodobieństwem proporcjonalnym do gdzie E jest energią stanu.

125
W celu zademonstrowania poprawności powyższego algorytmu rozważmy zmianę orientacji spinu i-tego atomu. Niech ta zmiana spowoduje przejście ze stanu o energii Ea do stanu o energii Eb przy czym Ea<Eb . Zgodnie z naszym algorytmem prawdopodobieństwo przejścia ze stanu a do stanu b wynosi natomiast prawdopodobieństwo zdarzenia odwrotnego jest równe: W równowadze termicznej zasada równowagi szczegółowej wymaga aby gdzie Pa i Pb są prawdopodobieństwami, że układ znajduje się w stanie a lub b. Powyższa zależność stwierdza, że w równowadze termicznej częstość przejść ze stanu a do stanu b jest taka sama jak ze stanu b do stanu a. Można ją przedstawić w następującej postaci: zgodnej z rozkładem Boltzmanna.

126
Niech każdy atom w naszej siatce ma 4 najbliższych sąsiadów, z wyjątkiem atomów brzegowych. Aby wyeliminować efekty brzegowe stosuje się periodyczne warunki brzegowe (tzn. zakłada się, że siatka rozpięta jest na powierzchni torusa). Wprowadźmy następującą definicję: Zgodnie z teorią średniego pola: Obliczenie pojemności cieplnej bezpośrednio ze wzoru jest trudne z uwagi na statystyczne fluktuacje energii E. Zamiast tego można skorzystać ze wzoru: gdzie E jest standardowym odchyleniem energii od wartości średniej E. Szczęśliwie, obliczanie E jest proste: wykorzystuje się standardowe odchylenia energii w kolejnych krokach symulacji Monte Carlo.

127
Na kolejnych rysunkach przedstawiono magnetyzację i pojemność cieplną w funkcji temperatury dla L= 5, 10, 20, i 40 przy zerowym polu zewnętrznym. W każdym przypadku wykonano 5000 iteracji. W obliczeniach średnich M, E, C pominięto 1000 pierwszych iteracji (aby układ mógł osiągnąć stan równowagi termicznej). Dla każdej wartości temperatury wyjściowa dwuwymiarowa siatka spinów była w pełni uporządkowana (zgodna orientacja spinów). Pod nieobecność zewnętrznego pola magnetycznego znak M nie ma znaczenia i dlatego na wszystkich wykresach M= M.
. Dla każdej wartości temperatury wyjściowa dwuwymiarowa siatka spinów była w pełni uporządkowana (zgodna orientacja spinów). Pod nieobecność zewnętrznego pola magnetycznego znak M nie ma znaczenia i dlatego na wszystkich wykresach M= M..")
128
Wypadkowa magnetyzacja M dwuwymiarowej siatki LxL=5x5 ferromagnetycznych atomów jako funkcja temperatury T/T0 przy zerowym zewnętrznym polu magnetycznym. Symulacje Monte-Carlo. Pojemność cieplna jako funkcja temperatury dla LxL=5x5. Linią ciągłą oznaczono pojemność cieplną obliczoną z równania: a przerywaną bezpośrednio z definicji:

129
Pojemność cieplna jako funkcja temperatury dla LxL=10x10.
Wypadkowa magnetyzacja M dwuwymiarowej siatki LxL=10x10 ferromagnetycznych atomów jako funkcja temperatury T/T0 przy zerowym zewnętrznym polu magnetycznym. Symulacje MC. Pojemność cieplna jako funkcja temperatury dla LxL=10x10.

130
Pojemność cieplna jako funkcja temperatury dla LxL=20x20.
Wypadkowa magnetyzacja M dwuwymiarowej siatki LxL=20x20 ferromagnetycznych atomów jako funkcja temperatury T/T0 przy zerowym zewnętrznym polu magnetycznym. Symulacje MC. Pojemność cieplna jako funkcja temperatury dla LxL=20x20.

131
Pojemność cieplna jako funkcja temperatury dla LxL=40x40.
Wypadkowa magnetyzacja M dwuwymiarowej siatki LxL=40x40 ferromagnetycznych atomów jako funkcja temperatury T/T0 przy zerowym zewnętrznym polu magnetycznym. Symulacje MC. Pojemność cieplna jako funkcja temperatury dla LxL=40x40.

132
Maksimum pojemności cieplnej (w jednostkach Nk) jako funkcja logarytmu z rozmiaru dwuwymiarowej siatki ferromagnetycznych atomów ln(L) pod nieobecność zewnętrznego pola magnetycznego. Symulacje Monte-Carlo. Zauważmy, że wysokość piku rośnie liniowo ze wzrostem logarytmu z rozmiaru układu, co sugeruje następującą relację:

133
Krzywe M(T), C(T) generowane w symulacjach Monte-Carlo wyglądają podobnie jak te uzyskane w ramach przybliżenia średniego pola. Podobieństwo rośnie wraz ze wzrostem rozmiaru układu symulacyjnego. Główną różnicą jest obecność resztkowej magnetyzacji powyżej temperatury krytycznej (czyli ogona) w symulacjach MC. Mówiąc inaczej, spontaniczne namagnesowanie nie maleje gwałtownie do zera gdy T > Tc i trwa jeszcze w pewnym zakresie temperatur. Wykresy pojemności cieplnej pokazują, że C obliczone wprost z definicji: wykazuje znaczne fluktuacje, natomiast C obliczone ze wzoru: charakteryzuje się małymi statystycznymi odchyleniami. Z tego względu ten drugi sposób obliczania C jest dużo lepszy. Należy zauważyć, że krzywe pojemności cieplnej mają kształt pików, tzn. pojemność cieplna nie jest równa zero dla T>Tc (dzięki resztkowej magnetyzacji).
.")
134
Najlepsze oszacowanie Tc uzyskane z maksimum piku C(T) wynosi 2. 27T0
Najlepsze oszacowanie Tc uzyskane z maksimum piku C(T) wynosi 2.27T0 . Temperatura krytyczna w ramach przybliżenia średniego pola to 2T0. Wynik symulacji jest zgodny z dokładnym rozwiązaniem otrzymanym w 1944 przez Onsagera dla dwuwymiarowego modelu Isinga: Analityczne rozwiązanie dwuwymiarowego modelu Isinga należy do najbardziej złożonych problemów fizyki teoretycznej i matematyki. Warto zauważyć, że dotychczas nie znaleziono rozwiązań analitycznych dla trzech i więcej wymiarów.
 wynosi 2.27T0 . Temperatura krytyczna w ramach przybliżenia średniego pola to 2T0. Wynik symulacji jest zgodny z dokładnym rozwiązaniem otrzymanym w 1944 przez Onsagera dla dwuwymiarowego modelu Isinga: Analityczne rozwiązanie dwuwymiarowego modelu Isinga należy do najbardziej złożonych problemów fizyki teoretycznej i matematyki. Warto zauważyć, że dotychczas nie znaleziono rozwiązań analitycznych dla trzech i więcej wymiarów.")
135
Dla układów fizycznych (makroskopowych) , (NA jest liczbą Avogadro)
Dla układów fizycznych (makroskopowych) , (NA jest liczbą Avogadro). Stąd C jest nieskończenie duże w temperaturze krytycznej (ponieważ ln(NA)>>1), jak to przedstawiono na rysunku poniżej (punkt osobliwy). Należałoby więc przedefiniować przejście fazowe drugiego rodzaju. Okazuje się bowiem, że tak naprawdę nieciągłość na krzywej pojemności cieplnej prawie nigdy nie występuje. Zamiast tego przejście fazowe drugiego rodzaju należałoby charakteryzować występowaniem punktu pozornie osobliwego na krzywej pojemności cieplnej. Przypomnimy, że zgodnie ze wzorem: Typowa amplituda fluktuacji energii jest proporcjonalna do pierwiastka kwadratowego z pojemności cieplnej tzn To oznacza, że amplituda fluktuacji energii staje się nieskończenie duża w sąsiedztwie punktu przejścia (temperatury krytycznej).
 , (NA jest liczbą Avogadro). Stąd C jest nieskończenie duże w temperaturze krytycznej (ponieważ ln(NA)>>1), jak to przedstawiono na rysunku poniżej (punkt osobliwy). Należałoby więc przedefiniować przejście fazowe drugiego rodzaju. Okazuje się bowiem, że tak naprawdę nieciągłość na krzywej pojemności cieplnej prawie nigdy nie występuje. Zamiast tego przejście fazowe drugiego rodzaju należałoby charakteryzować występowaniem punktu pozornie osobliwego na krzywej pojemności cieplnej. Przypomnimy, że zgodnie ze wzorem: Typowa amplituda fluktuacji energii jest proporcjonalna do pierwiastka kwadratowego z pojemności cieplnej tzn. . To oznacza, że amplituda fluktuacji energii staje się nieskończenie duża w sąsiedztwie punktu przejścia (temperatury krytycznej).")
136
W metodzie średniego pola problemem jest założenie, że każdy spin znajduje się w takim samym otoczeniu. Stąd, jeśli parametr wymiany J nie jest na tyle duży, aby spowodować całkowite uporządkowanie spinów atomowych, to uporządkowanie nie wystąpi wcale. To co obserwujemy w rzeczywistości powyżej temperatury krytycznej to zanik pełnego uporządkowania i występowanie jedynie uporządkowania lokalnego. Lokalne uporządkowane domeny mogą ulec zniszczeniu wskutek termicznych fluktuacji jeśli temperatura dostatecznie wzrośnie powyżej temperatury krytycznej. (Atomy w środku uporządkowanych domen znajdują się w innym otoczeniu niż atomy na obrzeżach, dlatego też przybliżenie średniego pola nie może przewidzieć istnienia domen).
..")
137
Obliczenia Monte-Carlo dla:
T=20T T=5T T=3T0 T=2.32T T=1.8T0 Magnetyzacja siatki 40x40 ferromagnetycznych atomów w równowadze termicznej, H=0. Czarne/białe kwadraciki oznaczają spiny zorientowane odpowiednio w kierunku plus/minus z.

138
Modele siatkowe – adsorpcja zlokalizowana
translacje x,y,z energia V – bariera dla dyfuzji V V kT<<V oscylacje x,y,z

139
MODEL LANGMUIRA - Zespół kanoniczny, N,T,V, B - const.
Dwuwymiarowa siatka B równocennych energetycznie centrów adsorpcyjnych. Adsorpcja zlokalizowana, monowarstwowa – co najwyżej jedna molekuła na jednym centrum adsorpcyjnym. Brak oddziaływań międzycząsteczkowych w fazie gazowej i zaadsorbowanej. N, V, T, B – ustalone parametry N-M - liczba molekuł w fazie gazowej w stanie równowagi siatka miejsc adsorpcyjnych M –liczba molekuł zaadsorbowanych w stanie równowagi

140
Poszukujemy zależności
W stanie równowagi dF=0 (F w minimum) lub dlnQc=0 (Qc-maksymalne) bo F=-kTlnQc Logarytmujemy Qc , stosujemy przybliżenie Stirlinga, skracamy.
 lub dlnQc=0 (Qc-maksymalne) bo. F=-kTlnQc. Logarytmujemy Qc , stosujemy przybliżenie Stirlinga, skracamy.")
141
=1 Ostatecznie mamy: Obliczamy pochodną lnQc po M:
i przyrównujemy ją do zera: Z równania stanu gazu doskonałego: =1 Ostatecznie mamy: lub

142
MODEL LANGMUIRA – Wielki zespół kanoniczny, ,T, B - const.
, T, B – ustalone parametry –liczba molekuł zaadsorbowanych w stanie równowagi Wyprowadzenie równania izotermy – poszukujemy lub w stałej T I. Wielka suma stanów: II. Logarytm z wielkiej sumy stanów: III. Średnia liczba cząsteczek w fazie zaadsorbowanej:

143
IV. Przejście od do W stanie równowagi: Dla gazu doskonałego: Podstawiając powyższe wyrażenie do równania na M() dostaniemy: gdzie:

153
STATYSTYKI KWANTOWE

154
Postulaty: W statystyce kwantowej, tak jak i w klasycznej, zakładamy, że średnie po zespole są równe średnim po czasie. Jako postulat przyjmujemy prawo jednakowych prawdopodobieństw stanów prostych, które mówi, że wszystkie dopuszczalne stany kwantowe o takiej samej energii są jednakowo prawdopodobne. Warunek ergodyczności układu przybiera następującą postać: Jeżeli układ, którego energia jest ustalona w bardzo wąskim przedziale, znajdował się początkowo w pewnym stanie kwantowym, to z upływem czasu będzie się znajdował kolejno we wszystkich stanach kwantowych o energii wewnątrz zadanego przedziału i to (średnio) przez jednakowy okres w każdym z nich. Teoretyczną granicę dokładności określenia energii układu stanowi zasada nieoznaczoności Heisenberga. Nieoznaczoność określenia energii układu jest odwrotnie proporcjonalna do czasu pełnej izolacji układu ( t): dla t=1s E=10-34J dla okresu pełnej izolacji t=1 rok (około 3•107 s) E=310-42J
 przez jednakowy okres w każdym z nich. Teoretyczną granicę dokładności określenia energii układu stanowi zasada nieoznaczoności Heisenberga. Nieoznaczoność określenia energii układu jest odwrotnie proporcjonalna do czasu pełnej izolacji układu ( t): dla t=1s E=10-34J. dla okresu pełnej izolacji t=1 rok (około 3•107 s) E=310-42J.")
155
Spin. Fermiony i bozony. Istnieją eksperymentalne dowody na to, że cząstki posiadają własny (mechaniczny) moment pędu (jeżeli cząstka jest naładowana, to z niezerowym mechanicznym momentem pędu jest związany niezerowy własny moment magnetyczny). Istnieją także cząstki nienaładowane, które mają niezerowy moment magnetyczny (np. neutron) Wartość własnego (spinowego) momentu pędu jest równa , spin s jest liczbą nieujemną, całkowitą lub połówkową, zależną od rodzaju cząstki. s=1/2 - spin większości cząstek elementarnych (elektronów, neutronów, protonów itp.) s= spin fotonu s= spin mezonu i mezonu K Rzut własnego momentu pędu cząstki na ustaloną oś z wynosi sz – liczba, która może dla danego s przybierać wartości: -s,-s+1,...,0,...,s+1,s. np. dla s=1 możliwe wartości sz wynoszą: -1,0,1 dla s=1/2 możliwe wartości sz wynoszą: -1/2,0,1/2
 moment pędu (jeżeli cząstka jest naładowana, to z niezerowym mechanicznym momentem pędu jest związany niezerowy własny moment magnetyczny). Istnieją także cząstki nienaładowane, które mają niezerowy moment magnetyczny (np. neutron) Wartość własnego (spinowego) momentu pędu jest równa , spin s jest liczbą nieujemną, całkowitą lub połówkową, zależną od rodzaju cząstki. s=1/2 - spin większości cząstek elementarnych (elektronów, neutronów, protonów itp.) s=1 - spin fotonu. s=0 - spin mezonu i mezonu K. Rzut własnego momentu pędu cząstki na ustaloną oś z wynosi. sz – liczba, która może dla danego s przybierać wartości: -s,-s+1,...,0,...,s+1,s. np. dla s=1 możliwe wartości sz wynoszą: -1,0,1. dla s=1/2 możliwe wartości sz wynoszą: -1/2,0,1/2.")
156
Wewnętrzny stan cząstki może różnić się wartością zmiennej sz
Wewnętrzny stan cząstki może różnić się wartością zmiennej sz . Aby określić kwantowo-mechaniczny stan cząstki, należy podać jej funkcję falową zależną od współrzędnych przestrzennych oraz liczbę sz . Dla cząstki poruszającej się w pudle potencjału należy podać cztery liczby kwantowe: nx,ny,nz (określające funkcję (x,y,z) ) oraz sz. Dla danej funkcji (x,y,z) możliwych jest 2s+1 stanów, różniących się tylko orientacją spinu. Pod nieobecność pola magnetycznego energia cząstki nie zależy od orientacji spinu (wartości sz) i występowanie zmiennej spinowej powoduje wzrost degeneracji każdego poziomu energetycznego 2s+1 razy - liczba stanów kwantowych o danej energii wzrasta 2s+1 razy. Liczba stanów własnych w określonym przedziale wartości energii cząstki poruszającej się w pudle potencjału wynosi: go =2s+1 - krotność degeneracji spinowej (każdej trójce liczb kwantowych nx,ny,nz odpowiada 2s+1 stanów kwantowych, różniących się jedynie orientacją spinu).
 ) oraz sz. Dla danej funkcji (x,y,z) możliwych jest 2s+1 stanów, różniących się tylko orientacją spinu. Pod nieobecność pola magnetycznego energia cząstki nie zależy od orientacji spinu (wartości sz) i występowanie zmiennej spinowej powoduje wzrost degeneracji każdego poziomu energetycznego 2s+1 razy - liczba stanów kwantowych o danej energii wzrasta 2s+1 razy. Liczba stanów własnych w określonym przedziale wartości energii cząstki poruszającej się w pudle potencjału wynosi: go =2s+1 - krotność degeneracji spinowej (każdej trójce liczb kwantowych nx,ny,nz odpowiada 2s+1 stanów kwantowych, różniących się jedynie orientacją spinu).")
157
Każdy punkt odpowiada zadanym nx, ny, nz
Każdy punkt odpowiada zadanym nx, ny, nz. Każdemu punktowi odpowiada go stanów różniących się składową spinu. Liczba mikrostanów o energii zawartej między E i E+dE wynosi:

158
Podział cząstek w zależności od tego, czy spin cząstki jest całkowity czy połówkowy:
BOZONY cząstki o spinie całkowitym (cząstki Bosego) np. foton, mezony i K FERMIONY cząstki o spinie połówkowym (cząstki Fermiego) np. e-, p, n, e+ Cząstki złożone - kwalifikacja do fermionów lub bozonów zależy od jej całkowitego spinu: Cząstka Bosego: składa się z parzystej liczby fermionów np. H, H2, 4He Cząstka Fermiego: składa się z nieparzystej liczby fermionów np. atom deuteru, cząsteczka HD. Funkcja falowa zespołu bozonów jest symetryczna, funkcja falowa zespołu fermionów - antysymetryczna względem przestawienia dowolnej pary cząstek. Fermiony podlegają zakazowi Pauliego.
 np. foton, mezony i K. FERMIONY cząstki o spinie połówkowym (cząstki Fermiego) np. e-, p, n, e+ Cząstki złożone - kwalifikacja do fermionów lub bozonów zależy od jej całkowitego spinu: Cząstka Bosego: składa się z parzystej liczby fermionów np. H, H2, 4He. Cząstka Fermiego: składa się z nieparzystej liczby fermionów np. atom deuteru, cząsteczka HD. Funkcja falowa zespołu bozonów jest symetryczna, funkcja falowa zespołu fermionów - antysymetryczna względem przestawienia dowolnej pary cząstek. Fermiony podlegają zakazowi Pauliego.")
159
Liczba sposobów rozmieszczenia 2 cząstek pomiędzy trzy stany kwantowe:
fermiony bozony cząstki klasyczne , - cząstki nieodróżnialne 1, 2 – cząstki odróżnialne - jednocząstkowy stan kwantowy Kombinacje bez powtórzeń Kombinacje z powtórzeniami Wariacje z powtórzeniami

160
Liczba sposobów realizacji rozkładu N cząstek pomiędzy poziomy energetyczne {1,2,…..}
i, gi 4, g4 3, g3 2, g2 Gdy cząstki są odróżnialne: 1, g1 Gdy cząstki są nieodróżnialne i są fermionami: Gdy cząstki są nieodróżnialne i są bozonami:

161
Warunek na ekstremum warunkowe:
Rozkład: Cząstki odróżnialne: M-B Cząstki nieodróżnialne - fermiony: F-D Cząstki nieodróżnialne - bozony: B-E Statystyki kwantowe przechodzą w statystykę klasyczną gdy Warunek ten jest spełniony w niezbyt niskich temperaturach i niezbyt wysokich ciśnieniach, a więc w spotykanych zazwyczaj warunkach fizykochemicznych. W bardzo niskich temperaturach oraz dla fotonów i elektronów musimy stosować statystyki kwantowe.

166
I. Podejście klasyczne Niech makroskopowy stan układu określa parametr X. Prawdopodobieństwo tego, że parametr X dla układu izolowanego (izolacja w sensie przybliżonym) ma wartość w przedziale od X do X+X wynosi. (X) – objętość fazowa (obszaru powłoki energetycznej) odpowiadająca mikrostanom, dla których wartość parametru X leży w przedziale <X; X+X>. Wiedząc, że dostaniemy tzn. prawdopodobieństwo określonego stanu makroskopowego układu izolowanego jest równe ilorazowi objętości fazowej odpowiadającej temu makrostanowi przez całkowitą objętość powłoki energetycznej.
 ma wartość w przedziale od X do X+X wynosi. (X) – objętość fazowa (obszaru powłoki energetycznej) odpowiadająca. mikrostanom, dla których wartość parametru X leży w przedziale <X; X+X>. Wiedząc, że dostaniemy. tzn. prawdopodobieństwo określonego stanu makroskopowego układu izolowanego. jest równe ilorazowi objętości fazowej odpowiadającej temu makrostanowi przez całkowitą objętość powłoki energetycznej.")
Podobne prezentacje